
| 제품명 |
|
|||
|---|---|---|---|---|
| 성분 / 함량 |
|
|||
| 첨가제 | ||||
| 도핑금지 약물정보 |
|
|||
| 전문 / 일반 |
|
단일 / 복합 | ||
| 제조 / 수입사 | ||||
| 제형 | 투여경로 | |||
| 성상 | ||||
| 허가일 |
|
|||
| 재심사 | ||||
| 대조 / 생동 | ||||
| 급여정보 |
|
|||
| ATC 코드 | ||||
| KPIC 약효분류 |
|
|||
| 제품설명서 | 보 기 ( 2017-10-10 게시 ) | |||
| 의약품안전성 정보(DUR) |
||||
| 포장단위 (식약처 기준) |
||||
| 저장방법 | ||||
사용자GNB바
컨텐츠
의약품 상세정보

허가정보 ∙ 복약정보
효능 · 효과
FLT3 ‑ITD 변이 양성인 새로 진단받은 급성 골수성 백혈병 성인 환자의 표준 시타라빈 및 안트라사이클린 유도요법과 표준 시타라빈 공고요법과의 병용 및 공고요법 후 단독 유지요법
용법 · 용량
이 약의 치료 시작 전에 FLT3 ‑ITD 변이 상태를 평가하여야 한다. FLT3 ‑ITD 변이 상태는 충분히 검증된 신뢰성 있는 시험방법을 사용하여 확인하여야 한다. 임상시험시 사용한 진단방법은 '사용상의 주의사항 5. 일반적 주의 1)항 및 14. 전문가를 위한 정보 3)항‘을 참조한다.
이 약은 표준 화학요법과 병용하여 각 유도요법 주기마다 2주간 35.4 mg 용량으로 1일 1회 투여한다. 완전관해(CR) 또는 불완전한 혈액학적 회복을 동반한 완전관해(CRi)에 도달한 환자의 경우, 이 약은 각 공고 화학요법 주기마다 2주간 35.4 mg을 1일 1회 투여한다. 이 약의 유지요법은 공고 화학요법 후 절대 중성구 수 >500/㎣ 및 혈소판 수 >50,000/㎣ 이상으로 회복되면 1일 1회 26.5 mg으로 시작한다. 2주 후, 프리데리시아 공식으로 보정한 QT 간격(QT interval corrected by Fridericia’s formula, QTcF)이 450 ms 이하인 경우, 유지 용량을 1일 1회 53 mg으로 증가한다(표 2 및 ‘3. 신중투여’ 항 참고). 유지 요법은 최대 36주기까지 계속할 수 있다.
추가적인 용량 및 용법에 대한 내용은 표 1 ‑ 3을 참고한다.
표 1: 이 약의 용법용량
|
이 약 시작 |
유도a |
공고b |
유지 |
|
제8일에 시작 (7+3 용법)c |
제6일에 시작 |
유지요법 제1일 | |
|
용량 |
1일 1회 35.4mg |
1일 1회 35.4mg |
ㆍ QTcF가 450 ms 이하인 경우 2주간 시작용량으로 1일 1회 26.5 mg 투여 ㆍ 2주 후, QTcF가 450 ms 이하인 경우, 1일 1회 53 mg 투여로 용량을 증가한다. |
|
기간 (28일 주기) |
각 주기마다 2주 |
각 주기마다 2주 |
최대 36주기 동안 주기 간에 휴약기간없이 1일 1회 투여 |
|
a 환자는 최대 2주기의 유도요법을 받을 수 있다. b 환자는 최대 4주기의 공고요법을 받을 수 있다. c 두 번째 유도 주기로서 투여되는 5 + 2 용법의 경우, 이 약은 제6일에 시작한다. | |||
이 약은 매일 거의 같은 시간에 경구 복용해야 하며 음식과 함께 또는 공복 상태에서 복용할 수 있다.
이 약은 통째로 삼켜야 하며, 씹거나 부수거나 분할해서는 안 된다.
2. 이상반응에 대한 모니터링 및 용량 조절
QTcF가 450 ms 이하인 경우에만 이 약 투여를 시작해야 한다(‘3. 신중투여’항 참고).
유도 및 공고요법 중, 치료 시작 전에 심전도(ECGs) 검사를 수행하고, 이후에는 이 약의 치료 중 주 1회 또는 임상적 증상에 따라 더 자주 수행해야 한다(‘3. 신중투여’항 참고).
유지요법 중, 치료 시작 전에 ECGs 검사를 수행하고, 투여 시작 및 용량 증가 후 처음 1개월 동안에는 주 1회 수행해야 하며, 그 후에는 임상적 증상에 따라 수행해야 한다. QTcF 간격이 450 ms 이하인 경우에만 용량을 증가해야 한다 (표 1 및 ‘3. 신중투여’항 참고).
전해질 이상(저칼륨혈증 및 저마그네슘혈증)은 교정해야 하며, 가능한 경우 QT 간격을 연장하는 약물의 병용투여를 피해야 한다 (‘3. 신중투여’항 참고).
이상반응에 따라 권장되는 용량 조절은 표 2를 참고한다. 이상반응 및/또는 강력한 CYP3A 억제제와 병용투여에 따른 용량 조절은 표 3을 참고한다.
표 2: 이상반응에 따른 용량 조절
|
이상반응 |
권장 조치 |
|
QTcF 450 - 480 ms (1등급) |
ㆍ 용량을 유지한다. |
|
QTcF 480 ms 초과 및 500 ms 이하 (2등급) |
ㆍ 투여를 일시 중단하지 않고 용량을 감소한다(표 3 참고). ㆍ QTcF가 450 ms 미만으로 회복되면 다음 주기에서 이전 용량으로 투여를 재개한다. 용량 증가 시 첫 주기 동안 QT 연장에 대해 면밀하게 모니터링한다. |
|
QTcF가 500 ms를 초과한 경우(3등급) |
ㆍ 이 약의 투여를 일시 중단한다. ㆍ QTcF가 450 ms 미만으로 회복되면 용량을 감소하여 투여를 재개한다 (표 3 참고). ㆍ 유도 및/또는 공고요법 중 QTcF의 500 ms 초과가 관찰되고 이 약과 관련 있는 것으로 의심되면, 유지 요법 중 1일 1회 53 mg 투여로 용량을 증가해서는 안 된다. 1일 1회 26.5 mg 용량을 유지한다. |
|
QTcF가 500 ms를 초과한 경우가 재발한 경우(3등급) |
ㆍ 적절한 용량 감소 및 다른 위험 요인(예: 혈청 전해질 이상, QT 간격을 연장시키는 약물 병용)의 교정/제거 조치를 취하였음에도 불구하고, QTcF가 500 ms를 초과한 경우가 재발한 경우, 이 약의 투여를 영구 중단한다. |
|
염전성 심실 빈맥 (Torsades de pointes), 여러 형태 심실 빈맥, 생명을 위협하는 부정맥의 징후/증상(4등급) |
ㆍ 이 약 투여를 영구 중단한다. |
|
3등급 또는 4등급의 비-혈액학적 이상반응 |
ㆍ 이 약의 투여를 일시 중단한다. ㆍ 이상반응이 1등급 이하로 개선되면, 이전 용량으로 투여를 재개한다. ㆍ 이상반응이 3등급 미만으로 개선되면, 용량을 감소하여 투여를 재개한다(표 3 참고). ㆍ 3등급 또는 4등급 이상반응이 28일 이상 지속되고 이 약과 관련 있는 것으로 의심되는 경우, 투여를 중단한다. |
|
활동성 골수 질환이 없는 지속성 4등급 중성구 감소증 또는 혈소판 감소증 |
ㆍ 용량을 감소한다(표 3 참고). |
|
이상반응은 National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03(NCI CTCAE v4.03)에 따른다. | |
표 3: 이 약으로 치료하는 동안 이상반응 및/또는 강력한 CYP3A 억제제와 병용투여에 따른 단계별 용량 조절
|
치료 단계 |
전체 용량 |
용량 감소 | ||
|
이상반응 발생 |
강력한 CYP3A 억제제와 병용투여 |
이상반응 발생 및 강력한 CYP3A 억제제와 병용투여 | ||
|
유도 또는 공고 |
35.4 mg |
26.5 mg |
17.7 mg |
투여 일시 중단 |
|
유지 (처음 2주) |
26.5 mg |
투여 일시 중단 |
17.7 mg |
투여 일시 중단 |
|
유지 (2주 후) |
53 mg |
35.4 mg |
26.5 mg |
17.7 mg |
4. 복용 누락 또는 구토
이 약의 복용을 누락하였거나 예정 시간에 복용을 하지 못한 경우, 가능한 빨리 같은 날에 복용하여야 하고, 다음 복용일에 원래의 일정대로 복용하여야 한다. 하루에 2회 용량을 복용하지 않아야 한다.
이 약 복용 후 구토가 있는 경우, 당일 추가 용량을 복용하지 않아야 한다. 다음 복용일에 원래의 일정대로 복용해야 한다.
사용상의 주의사항
1. 경고 (‘3. 신중투여’항 참고)
이 약으로 치료받은 환자에서 염전성 심실 빈맥(torsades de pointes), 심실세동, 심정지 및 급사가 발생했다.
임상시험에서 이 약으로 치료받은 급성 골수성 백혈병 환자 1,081명 중 약 0.1 %에서 염전성 심실 빈맥(torsades de pointes)이 발생했고, 0.6 %에서 심정지가 발생했으며 그 중 0.4 %는 치명적인 결과를 나타냈고, 0.1 %의 환자에서 심실세동이 발생했다. 이러한 중증 부정맥은 주로 유도 단계에서 발생했다.
2. 다음 환자에는 투여하지 말 것
1) 이 약의 주성분 또는 구성성분에 과민증이 있는 환자
2) QT 연장 증후군, 심실성 부정맥 또는 염전성 심실 빈맥(torsades de pointes) 병력, 중증 저칼륨혈증, 중증 저마그네슘혈증이 있는 환자(‘3. 신중투여’항 참고).
3. 다음 환자에는 신중히 투여할 것
1) QT 간격 연장
이 약은 QT 간격 연장과 관련이 있다. QT 간격 연장은 심실성 부정맥 또는 염전성 심실 빈맥(torsades de pointes)의 위험성을 증가시킬 수 있다. 선천성 QT 연장 증후군 및/또는 염전성 심실 빈맥(torsades de pointes)의 병력이 있는 환자는 퀴자티닙의 개발 프로그램에서 제외되었다. QT 연장 증후군, 심실성 부정맥 또는 염전성 심실 빈맥(torsades de pointes) 병력, 중증 저칼륨혈증, 중증 저마그네슘혈증이 있는 환자에게 이 약을 투여해서는 안 된다.
이 약은 QT 간격 연장이 발생할 유의한 위험이 있는 환자에게 주의해서 사용해야 한다. 이는 조절되지 않거나 유의한 심혈관 질환(예. 2도 또는 3도 심장차단(박동조율기 없이), 6개월 이내 심근경색, 조절되지 않는 협심증, 조절되지 않는 고혈압), 울혈성 심부전 병력이 있는 환자 및 QT 간격을 연장시키는 것으로 알려진 약물을 병용 투여한 환자를 포함한다. 전해질은 정상 범위로 유지되어야 한다 (‘용법용량’ 참조).
이 약의 치료 시작 전 ECGs 검사를 수행해야 하고, 전해질 이상이 있는 경우 투여 시작 전 교정하여야 한다. QTcF 간격이 450 ms 초과인 경우, 이 약 투여를 시작해서는 안 된다.
유도 및 공고요법 중, ECGs 검사는 투여 시작 전 수행하고, 이후에는 이 약 투여 중 주 1회 또는 임상적 증상에 따라 더 자주 수행해야 한다.
유지요법 중, ECGs 검사는 투여 시작 전 수행하고, 투여 시작 및 용량 증가 후 처음 1개월 동안에는 주 1회 수행해야 하며, 그 후에는 임상적 증상에 따라 수행해야 한다. QTcF 간격이 450 ms 초과인 경우 유지 시작 용량을 증가해서는 안 된다(표 1 참고).
생명을 위협하는 부정맥의 징후 또는 증상과 함께 QT 간격 연장이 나타난 환자에 대해서는 이 약을 영구 중단한다(‘용법 ▪용량’ 참고).
QT 간격의 ECG 모니터링은 QT 간격 연장 및 염전성 심실 빈맥(torsades de pointes)이 발생할 위험이 유의하게 있는 환자에서 더 자주 수행되어야 한다.
이 약의 치료 시작 전 및 치료 중에 저칼륨혈증 및 저마그네슘혈증의 모니터링 및 교정을 수행해야 한다. 설사나 구토를 경험한 환자에서는 전해질 및 심전도를 더 자주 모니터링 해야 한다.
⓵ QT 간격을 연장하는 약물 투여 시 ECG 모니터링
이 약과 QT 간격을 연장하는 것으로 알려진 약물을 함께 투여하는 경우, 환자의 ECG를 더 자주 모니터링 해야 한다. QT 간격을 연장하는 약물의 예는 아졸계 항진균제, 온단세트론, 그라니세트론, 아지스로마이신, 펜타미딘, 독시사이클린, 목시플록사신, 아토바쿠온, 프로클로르페라진 및 타크로리무스가 포함되지만 이에 국한되지 않는다.
⓶ 강력한 CYP3A 억제제와 병용 투여
이 약과 강력한 CYP3A 억제제를 병용 투여하는 경우, 퀴자티닙의 노출을 증가시킬 수도 있으므로 이 약의 용량을 감소해야 한다 (‘용법 ▪용량’ 참고).
2) 배 ▪태아 독성
동물 시험 결과에 근거하여, 이 약은 임부에게 투여 시 배 ▪태아에 위해할 수 있다. 임부에게 태아에 대한 잠재적 위험에 대해 알린다. 임신 가능성이 있는 여성에게 이 약 투여 중 및 마지막 투여 이후 최소 7개월 동안 효과적인 피임법을 사용해야 한다. 임신 가능성이 있는 배우자가 있는 남성은 이 약 투여 중 및 마지막 투여 이후 최소 4개월 동안 효과적인 피임법을 사용해야 한다.(‘7. 임부 및 수유부에 대한 투여’항 참고).
4. 이상반응
1) 안전성 프로파일 요약
이 약의 안전성은 FLT3 ‑ITD 양성인 새로 진단받은 급성 골수성 백혈병 성인 환자를 대상으로 한 무작위배정, 이중 눈가림, 위약 대조 임상시험(QuANTUM ‑First)에서 조사되었다. 환자들은 이 약(N=265) 또는 위약(N=268)을 표준 화학요법과 병용 투여받거나 단독 유지요법으로 투여받았다(‘14. 전문가를 위한 정보’항 참고). 전체 치료 기간의 중앙값은 이 약 치료군의 환자에 대해서는 10.7주(범위: 0.1 ~ 184.1)였고 위약 치료군의 환자에 대해서는 9.5주(범위: 0.4 ~ 181.9)였다.
전체 시험기간 중, 이 약 치료군에서 가장 흔하게 보고된 이상반응(발생률 >20%)은 알라닌 아미노 전이 효소 증가(58.9%), 혈소판 수 감소(40.0%), 헤모글로빈 감소(37.4%), 설사(37.0%), 구역(34.0%), 복통(29.4%), 두통(27.5%), 구토(24.5%) 및 중성구 수 감소(21.9%)였다. 가장 흔하게 보고된 3등급 또는 4등급 이상반응(발생률 ≧5%)은 혈소판 수 감소(40.0%), 헤모글로빈 감소(35.5%), 중성구 수 감소(21.5%), 알라닌 아미노 전이 효소 증가(12.1%) 균혈증(7.2%) 및 진균 감염(5.7%)이었다.
이 약 치료군에서 가장 흔하게 보고된 중대한 이상반응(발생률 ≧2%)은 중성구 감소증(3.0%), 진균 감염(2.3%) 및 헤르페스 감염(2.3%)이었다. 치명적인 결과를 나타낸 이상반응은 진균 감염(0.8%) 및 심정지(0.4%)였다.
이 약의 투여 일시 중단과 연관된 가장 흔하게 보고된 이상반응(발생률 ≧2%)은 중성구 감소증(10.6%), 혈소판 감소증(4.5%) 및 심전도 QT 연장(2.6%)이었다. 이 약의 용량 감소와 연관된 가장 흔하게 보고된 이상반응(발생률 ≧2%)은 중성구 감소증(9.1%), 혈소판 감소증(4.5%) 및 심전도 QT 연장(3.8%)이었다.
이 약의 영구 중단과 연관된 가장 흔하게 보고된 이상반응(발생률 ≧1%)은 혈소판 감소증(1.1%)이었다.
단독 유지요법 중 안전성 프로파일
유지 단계에 진입한 208명(39%)의 환자는 이 약(116명) 또는 위약(92명)을 투여받았다. 유지 요법을 위한 치료기간의 중앙값은 이 약 치료군의 환자에 대해서는 67.4주(범위: 0.4 ~ 167.0)였고 위약 치료군의 환자에 대해서는 67.7주(범위: 0.3 ~ 167.4)였다.
이 약 치료군에서 가장 흔하게 보고된 이상반응은 중성구 수 감소(87.9%), 혈소판 수 감소(72.4%), 헤모글로빈 감소(55.2%), 알라닌 아미노 전이 효소 증가(49.1%), 상기도 감염(27.6%), 구역(23.3%) 및 설사(20.7%)였다. 이 약 투여 시 가장 빈번하게 보고된 3등급 또는 4등급 이상반응은 중성구 수 감소(71.6%), 혈소판 수 감소(24.1%) 및 헤모글로빈 감소(18.1%)였다.
이 약 치료군에서 가장 흔하게 보고된 중대한 이상반응은 헤르페스 감염(3.4%), 중성구 감소증(2.6%), 상기도 감염(1.7%) 및 구토(1.7%)였다. 유지 요법 중 치명적인 이상반응은 발생하지 않았다.
이 약의 투여 일시 중단과 연관된 가장 흔하게 보고된 이상반응은 중성구 감소증(24.1%), 혈소판 감소증(10.3%) 및 심전도 QT 연장(4.3%)이었다. 이 약의 용량 감소와 연관된 가장 흔하게 보고된 이상반응은 중성구 감소증(20.7%), 혈소판 감소증(10.3%) 및 심전도 QT 연장(5.2%)이었다. 이 약의 영구 중단과 연관된 가장 흔하게 보고된 이상반응은 혈소판 감소증(2.6%), 구역, 중성구 감소증 및 식욕 감소(각각 1.7%)였다.
2) 이상반응 표
이상반응은 MedDRA 기관계 대분류(SOC)에 따라 기재되었다. 각 기관계 내에서, 이상반응은 다음의 통상의 규칙(CIOMS III)을 이용하여 가장 빈도가 높은 반응 순으로 빈도별로 정렬하였다: 매우 흔하게( ≧1/10), 흔하게( ≧1/100 ~ <1/10), 흔하지 않게( ≧1/1000 ~ <1/100), 드물게( ≧1/10,000 ~ <1/1000), 매우 드물게(<1/10,000). 각 빈도 범주 내에서 이상반응은 중증도가 감소하는 순서대로 기재하였다.
표 4: 이 약의 임상시험(QuANTUM ‑First)에서 관찰된 이상반응
|
약물이상반응 |
이 약 + 표준 화학요법 N=265 |
위약 + 표준 화학요법 N=268 | ||
|
모든 등급 (%) |
3등급 또는 4등급 (%) |
모든 등급 (%) |
3등급 또는 4등급 (%) | |
|
감염 및 침습 | ||||
|
매우 흔하게 | ||||
|
상기도 감염a |
18.1 |
1.9 |
10.1 |
2.2 |
|
진균 감염b |
15.1 |
5.7 |
9.7 |
3.0 |
|
헤르페스 감염c |
14.0 |
3.0 |
8.2 |
1.5 |
|
균혈증d |
11.3 |
7.2 |
4.1 |
3.7 |
|
혈액 및 림프계 이상 | ||||
|
흔하게 | ||||
|
범혈구 감소증 |
2.6 |
2.3 |
0.4 |
0.4 |
|
매우 흔하게 | ||||
|
혈소판 감소증e |
40.0 |
40.0 |
44.0 |
44.0 |
|
빈혈e |
37.4 |
35.5 |
45.5 |
42.5 |
|
중성구 감소증e |
21.9 |
21.5 |
21.6 |
20.1 |
|
대사 및 영양 이상 | ||||
|
매우 흔하게 | ||||
|
식욕 감소 |
17.4 |
4.9 |
13.4 |
1.9 |
|
신경계 이상 | ||||
|
매우 흔하게 | ||||
|
두통f |
27.5 |
0 |
19.8 |
0.7 |
|
심장 이상 | ||||
|
흔하지 않게 | ||||
|
심정지g |
0.8 |
0.4 |
0 |
0 |
|
심실세동g |
0.4 |
0.4 |
0 |
0 |
|
호흡기, 흉곽 및 종격 이상 | ||||
|
매우 흔하게 | ||||
|
비출혈 |
15.1 |
1.1 |
10.8 |
0.4 |
|
위장관 이상 | ||||
|
매우 흔하게 | ||||
|
설사h |
37.0 |
3.8 |
35.1 |
3.7 |
|
구역 |
34.0 |
1.5 |
31.3 |
1.9 |
|
복통i |
29.4 |
2.3 |
22.0 |
1.1 |
|
구토 |
24.5 |
0 |
19.8 |
1.5 |
|
소화 불량 |
11.3 |
0.4 |
8.6 |
0.7 |
|
간담도계 이상 | ||||
|
매우 흔하게 | ||||
|
알라닌 아미노 전이 효소 증가e |
58.9 |
12.1 |
60.1 |
10.4 |
|
전신 이상 및 투여 부위 상태 | ||||
|
매우 흔하게 | ||||
|
부종j |
18.9 |
0.4 |
19.0 |
1.5 |
|
임상 검사 | ||||
|
매우 흔하게 | ||||
|
심전도 QT 연장k |
14.0 |
3.0 |
4.1 |
1.1 |
|
표준 화학요법 = 시타라빈(사이토신 아라비노시드) 및 안트라사이클린(다우노루비신 또는 이다루비신) a 상기도 감염은 상기도 감염, 비인두염, 부비동염, 비염, 편도염, 인후두염, 세균성 인두염, 인두 편도염, 바이러스 인두염 및 급성 부비동염을 포함함. b 진균 감염은 구강 칸디다증, 기관지 폐 아스페르길루스증, 진균 감염, 외음질 칸디다증, 아스페르길루스 감염, 진균성 하기도 감염, 진균성 구강 감염, 칸디다 감염, 진균성 피부 감염, 털곰팡이증, 구강 인두 칸디다증, 구강 아스페르길루스증, 진균성 간 감염, 간 비장 칸디다증, 손발톱 진균증, 진균 혈증, 전신 칸디다 및 전신 진균증을 포함함. c 헤르페스 감염은 구강 헤르페스, 대상 포진, 헤르페스 바이러스 감염, 단순 포진, 사람 헤르페스 바이러스 6형 감염, 생식기 헤르페스 및 헤르페스 피부염을 포함함. d 균혈증은 균혈증, 클레브시엘라 균혈증, 포도상 구균 균혈증, 장구균성 균혈증, 연쇄구균 균혈증, 기기 관련 균혈증, 대장균 균혈증, 코리네박테륨 균혈증 및 슈도모나스 균혈증을 포함함. e 용어들은 실험실 데이터를 기반으로 한다. f 두통은 두통, 긴장성 두통 및 편두통을 포함함. g 1명이 2건의 사례(심실세동 및 심정지)를 경험하였다. h 설사는 설사 및 출혈성 설사를 포함함. i 복통은 복통, 상복부 통증, 복부 불편감, 하복부 통증 및 위장관 통증을 포함함. j 부종은 말초 부종, 안면 부종, 부종, 체액 과부하, 전신 부종, 말초 종창, 국소 부종 및 얼굴 종창을 포함함. k 심전도 QT 연장은 심전도 QT 연장 및 심전도 QT 간격 이상을 포함함. | ||||
이 약은 ECG에서 QT 간격을 연장한다. 등급에 관계없이, 치료 후 발생한 이상반응으로써 QT 간격 연장은 이 약을 투여한 환자의 14.0%에서 보고되었고, 환자의 3.0%가 3등급 이상의 이상반응을 경험하였다. QT 연장은 환자 10명(3.8%)에서는 용량 감소, 7명(2.6%)에서는 투여 일시 중단, 2명(0.8%)에서는 영구 중단을 초래하였다. 중앙에서 검토된 ECG 데이터에 근거하여, QTcF >500 ms는 환자의 2.3%에서 발생하였다. 이 약을 투여한 환자 2명(0.8%)이 심실세동 기록과 함께 심정지를 경험하였고, 1명은 치명적인 결과를 나타냈으며, 이들 모두 중증 저칼륨혈증이 있는 상황에서 발생하였다. 이 약 투여 전 및 투여 중 심전도 검사 및 모니터링, 저칼륨혈증 및 저마그네슘혈증 교정을 실시해야 한다. QT 간격 연장이 있는 환자에 대한 이 약의 용량 조절은 ‘용법 ▪용량’ 항을 참고한다.
5. 일반적 주의
1) FLT3 ‑ITD 변이 검사
이 약은 FLT3 ‑ITD 변이 양성인 급성 골수성 백혈병 환자에서만 치료 이익을 보였으므로, 이 약의 치료를 받을 환자를 선택하기 위해서는 FLT3 ‑ITD 변이 양성인 급성 골수성 백혈병의 진단이 필요하다. FLT3 ‑ITD 상태는 임상시험 분석법(PCR과 모세관 전기영동법)을 사용하여 전향적으로 확인되었고, FDA에서 승인받은 분석법인 LeukoStratⓡ CDx FLT3 분석 동반진단기기를 사용하여 후향적으로 검증되었다.
6. 약물 상호작용
1) 강력한 CYP3A 억제제
건강한 대상자에게 이 약과 강력한 CYP3A 억제제와 병용투여 시, 이 약 단독 투여와 비교하여 퀴자티닙의 노출이 증가하였다. 이 약의 1회 투여 용량(26.5 mg)과 케토코나졸(200 mg씩 1일 2회)을 병용투여 시 퀴자티닙의 Cmax는 17 %, AUCinf는 94 % 증가하였다. 활성 대사체인 AC886의 AUCinf는 15 %, Cmax는 60 % 감소했다. 정상 상태에서 퀴자티닙의 노출(Cmax 및 AUC0 ‑24h)은 각각 86 %, 96 % 증가할 것으로 추정되었다. 퀴자티닙의 노출 증가로 독성의 위험성이 증가할 수도 있다.
강력한 CYP3A 억제제와의 병용투여가 불가피한 경우, 이 약의 용량을 감소해야 한다(‘용법용량’ 참고). 강력한 CYP3A 억제제의 예로 케토코나졸, 이트라코나졸, 포사코나졸, 보리코나졸, 클래리스로마이신, 네파조돈, 텔리스로마이신 및 항레트로바이러스제가 있다.
2) 강력한 및 중등도의 CYP3A 유도제
건강한 대상자에게 이 약과 중등도의 CYP3A 유도제의 병용투여 시, 이 약 단독 투여(총 53 mg)와 비교하여 퀴자티닙 및 활성 대사체 AC886의 노출이 감소하였다. 중등도의 CYP3A 유도제인 에파비렌즈(600 mg 씩 1일 1회)와 병용투여 시 퀴자티닙 Cmax 및 AUCinf는 각각 약 45 %와 90 % 감소하였다. AC886의 Cmax 및 AUCinf는 각각 약 68 % 및 96 % 감소하였다 (‘14. 전문가용 정보’항 참고).
퀴자티닙 노출 감소는 유효성 감소로 이어질 수 있다. 이 약과 강력한 또는 중등도의 CYP3A 유도제와의 병용투여를 피해야 한다.
강력한 CYP3A4 유도제의 예로는 아팔루타마이드, 카르바마제핀, 엔잘루타마이드, 미토탄, 페니토인, 리팜피신, 세인트 존스 워트(Hypericum perforatum 라고도 함) 등 특정 생약 제제가 있다. 중등도의 CYP3A4 유도제의 예로는 에파비렌즈, 보젠탄, 에트라비린, 페노바르비탈 및 프리미돈이 있다.
3) 수송체
⓵ P ‑당단백(P ‑gp) 기질
퀴자티닙과 다비가트란 에텍실레이트(P ‑gp 기질)를 병용투여하였을 때, 총 및 유리 다비가트란의 Cmax는 각각 12% 및 13% 증가하였고, AUCinf는 각각 13% 및 11% 증가하였다(‘14. 전문가용 정보’항 참고). 퀴자티닙은 약한 P ‑gp 억제제이며, 이 약과 P ‑gp 기질을 병용투여 시 용량 조절은 권장되지 않는다.
⓶ 유방암 내성 단백질(BCRP) 기질
퀴자티닙은 in vitro IC50 추정값 0.813 μM에서 유방암 내성 단백질을 저해한다. 생리학적 기반 약동학(PBPK) 분석에 따르면 퀴자티닙 53mg은 로수바스타틴의 AUC0 ‑48h를 2.44배, 설파살라진은 2.66배 증가시킬 것으로 예측되었다. 퀴자티닙을 BCRP 기질 약물과 병용투여할 경우 주의해야 한다. BCRP 기질 약물의 용량 감소를 고려해야 한다.
⓷ Uridine Diphosphate Glucuronosyltransferases(UGT) 1A1 기질
퀴자티닙은 in vitro Ki 추정값 0.78 μM에서 UGT1A1을 저해한다. PBPK 분석에 근거하여, 퀴자티닙은 랄테그라비르(UGT1A1 기질)의 Cmax와 AUCinf를 3% 증가시킬 것으로 예상되었다. UGT1A1 기질과 이 약의 병용투여 시 용량 조절은 권장되지 않는다.
⓸ QT 간격을 연장시키는 약물
이 약과 QT 간격을 연장하는 기타 약물의 병용투여는 QT 연장의 발생률을 더욱 증가시킬 수 있다. 이 약과 QT 간격을 연장하는 약물을 병용투여 시 주의해야 한다(‘3. 다음 환자에는 신중히 투여할 것 중 ‘1) QT 간격 연장’항 참고).
7. 임부 및 수유부에 대한 투여
1) 남성 및 여성의 피임
이 약은 임부에게 투여 시 배 ▪태아에 해를 끼칠 수 있다(‘14. 전문가용 정보’항 참고). 잠재적인 임신 가능성이 있는 여성은 이 약 투여 중 및 마지막 투여 후 최소 7개월 동안 효과적인 피임법을 사용하여야 한다. 임신 가능성이 있는 여성 배우자가 있는 남성은 이 약 투여 중 및 마지막 투여 후 최소 4개월 동안 효과적인 피임법을 사용하여야 한다.
2) 임부
임부에게 퀴자티닙을 투여한 자료는 없다. 동물시험 결과에 근거하여, 이 약은 임부에게 투여 시 배 ▪태아 독성을 초래할 수 있다 (‘14. 전문가용 정보’항 참고).
이 약을 임신 중 투여해서는 안 된다. 임부에게 태아에 대한 잠재적 위험에 대해 알린다.
3) 수유부
퀴자티닙 또는 활성 대사체가 모유로 분비되는지 여부는 알려져 있지 않다. 모유 수유 중인 소아에서 중대한 이상반응이 발생할 가능성이 있기 때문에, 이 약 투여 중 및 마지막 투여 후 최소 5주 동안 모유 수유를 해서는 안 된다.
4) 임신 가능성이 있는 여성
임신 가능성이 있는 여성은 이 약으로 치료를 시작하기 전 임신 검사를 받아야 한다.
5) 생식능
이 약이 사람의 생식능에 미치는 영향에 대한 자료는 없다. 동물 시험 결과에 근거할 때, 이 약 투여로 인해 여성 및 남성 생식능이 손상될 수 있다(‘14. 전문가를 위한 정보’항 참고).
8. 소아에 대한 투여
이 약은 18세 미만의 소아 및 청소년에서 안전성 및 유효성이 확립되지 않았다. 가용할 수 있는 데이터가 없다.
9. 고령자에 대한 투여
65세 이상 환자에서 이 약의 용량 조절은 필요하지 않다.
임상시험(QuANTUM ‑First)에서 이 약을 투여받은 새로 진단받은 급성 골수성 백혈병 환자 265명 중 69명(26%)이 65세 이상이었고 1명(0.4%)이 75세였다. 65세 이상 환자와 젊은 성인 환자 간에 안전성에서 전반적인 차이는 관찰되지 않았다.
이 약의 임상시험에는 젊은 성인 환자와 다르게 반응을 보이는지를 확인하기에 충분한 수의 65세 이상 환자가 포함되지 않았다.
이 약은 특히 치료 초기 단계에서 젊은 환자에 비해 고령 환자(즉, 65세 이상)에서 치명적인 감염 발생 빈도가 더 높았다. 65세 이상 환자는 유도 요법 기간 동안 중증 감염 발생 여부를 면밀히 모니터링해야 한다.
10. 신 장애 환자에 대한 투여
경증 또는 중등도 신 장애 환자에서 용량 조절은 권장되지 않는다.
이 약은 중증 신 장애(Cockcroft ‑Gault 식으로 추정한 CLcr <30 mL/min) 환자에서 안전성 및 유효성이 평가되지 않았기 때문에, 중증 신 장애 환자에게는 권장되지 않는다.
11. 간 장애 환자에 대한 투여
경증 또는 중등도 간 장애 환자에서 용량 조절은 권장되지 않는다.
이 약은 중증 간 장애(Child ‑Pugh Class C 또는 총 빌리루빈 >3 x ULN 및 모든 수치의 AST) 환자에서 안전성 및 유효성이 평가되지 않았기 때문에, 중증 간 장애 환자에게는 권장되지 않는다.
12. 과량투여시의 처치
이 약의 과량투여에 대한 해독제는 알려지지 않았다. 과량투여 시 투여를 일시 중단하고, QT 간격 연장을 측정하기 위한 ECG를 수행하며, 적절한 보조 조치를 한다(‘용법용량’ 및 ‘3. 신중투여’항 참고).
13. 보관 및 취급 시 주의사항
1) 이 약은 어린의 손이 닿지 않는 곳에 보관해야 한다.
2) 이 약을 다른 용기에 옮겨 담지 않는다.
14. 전문가를 위한 정보
1) 약리작용
(1) 작용 기전
퀴자티닙은 티로신 키나아제 FLT3 수용체의 저분자 억제제다. 퀴자티닙 및 그 주요 순환 활성 대사체 AC886은 FLT3의 아데노신삼인산(ATP) 결합 부위에 높은 친화성(각각 Kd = 1.3 nM 및 0.54 nM)으로 경쟁적으로 결합한다. 퀴자티닙 및 AC886은 수용체의 자가인산화를 방지하여 FLT3 키나아제 활성을 억제하며, 이후 다운스트림 FLT3 수용체 신호전달을 억제하고 FLT3 ‑ITD 의존적인 세포 증식을 차단한다.
(2) 약력학적 효과
심장 전기생리학
임상시험(QuANTUM ‑First)의 노출 ‑반응 분석에서 유지요법 중 퀴자티닙(53 mg)의 정상 상태 Cmax에서 연장된 QTcF 간격은 24.1 ms(양측 90% CI의 상한: 26.6 ms)였으며 농도 ‑의존적인 QTcF 간격 연장이 예상되었다.
2) 약동학적 특성
퀴자티닙 및 주요 순환 활성 대사체(AC886)의 약동학은 건강한 성인과 성인 암 환자에서 분석되었으며, 달리 명시되지 않는 한 기하 평균(변동계수 [%CV])으로 제시되었다.
급성 골수성 백혈병 성인 환자에서 1일 1회 투여 후 15일 째에 퀴자티닙 농도가 정상 상태(steady state)에 도달했다. 건강한 성인의 경우 퀴자티닙 노출(AUC 및 Cmax)은 26.5 mg (권장 유도 용량 35.4 mg의 0.75배)에서 79.5 mg (권장 유도 용량 35.4 mg의 2.25배)까지, 급성 골수성 백혈병 성인 환자의 경우 17.7mg (권장 유도 용량 35.4mg의 0.5배)에서 53 mg (권장 유도 용량 35.4 mg의 1.5배)까지의 용량 범위에서 비례적으로 증가했다.
표 5. 퀴자티닙 및 AC886의 약동학적 특성
|
|
퀴자티닙 |
AC886 |
|
새로 진단된 급성 골수성 백혈병 환자의 유도 요법 중 이 약 35.4mg을 1일 1회 투여한 후 정상 상태 노출도 | ||
|
Cmax |
140 ng/mL (71%) |
163 ng/mL (52%) |
|
AUC0-24h |
2,680 ng·h/mL (85%) |
3,590 ng·h/mL (51%) |
|
새로 진단된 급성 골수성 백혈병 환자의 공고 요법 중 이 약 35.4mg을 1일 1회 투여한 후 정상 상태 노출도 | ||
|
Cmax |
204 ng/mL (64%) |
172 ng/mL (47%) |
|
AUC0-24h |
3,930 ng·h/mL (78%) |
3,800 ng·h/mL (46%) |
|
새로 진단된 급성 골수성 백혈병 환자의 유지 요법 중 이 약 53mg을 1일 1회 투여한 후 정상 상태 노출도 | ||
|
Cmax |
529 ng/mL (60%) |
262 ng/mL (48%) |
|
AUC0-24h |
10,200 ng·h/mL (75%) |
5,790 ng·h/mL (46%) |
|
축적비* (AUC0-24h) |
5.4 (4.4) |
8.7 (6.8) |
(1) 흡수
정제 제형에서 퀴자티닙의 절대 생체이용률은 71 %였다. 건강한 성인에게 공복 상태에서 경구 투여 시 퀴자티닙 및 AC886의 최고 농도 도달 시간(Tmax 중앙값)은 각각 약 4시간(범위 2 ‑ 8시간) 및 5 ‑ 6시간(범위 4 ‑ 120시간)이었다. 53 mg/일 용량으로 유지요법 중 정상 상태에서, 퀴자티닙 및 AC886의 기하평균(%CV) Cmax는 각각 529 ng/mL(60 %) 및 262 ng/mL(48 %)로 추정되었고, 기하평균(%CV) AUC0 ‑24h는 각각 10,200 ng ▪h/mL(75 %) 및 5,790 ng ▪h/mL(46 %)였다.
음식과 함께 이 약 투여 시 퀴자티닙 Cmax는 8% 감소하였으며, AUCinf는 8% 증가하였다. Tmax는 2시간 지연되었다. 이 약은 음식 여부와 관계없이 투여 가능하다.
(2) 분포
퀴자티닙 및 AC886의 In vitro 사람 혈장 단백질 결합률은 각각 99% 이상이다.
10 ‑ 1,000 ng/mL 농도 범위에서 in vitro 적혈구 내 퀴자티닙 및 AC886의 혈액 대 혈장 비율은 각각 0.79 ‑ 1.30 및 1.36 ‑ 3.19이다.
건강한 성인에서 퀴자티닙의 정상 상태에서 분포 용적(Vss)의 기하평균(%CV)은 275 L(17%)로 추정되었다.
(3) 생체 내 변환
퀴자티닙은 활성 대사체 AC886을 생성하는 산화 경로를 통해 in vitro에서 CYP3A4/5에 의해 주로 대사되며, 이후 CYP3A4/5에 의해 추가로 대사된다. 유지요법 중 정상 상태에서 퀴자티닙에 대한 AC886의 AUC0 ‑24h비는 0.57이었다.
(4) 배설
퀴자티닙 및 AC886의 유효 반감기(t1/2)의 평균(표준편차)는 새로 진단된 급성 골수성 백혈병 환자에서 각각 81시간(±73) 및 136시간(±113)이다. 퀴자티닙 및 AC886의 축적비의 평균 AUC0 ‑24h(표준편차)는 각각 5.4(±4.4) 및 8.7(±6.8)이었다.
퀴자티닙 및 그 대사체는 대부분이 일차적으로 간담도 경로를 통해 제거되어 대변으로 배설된다(경구 투여된 방사성 동위원소 표지 투여량의 76.3 %). 대변으로 배설되는 미변화체 퀴자티닙은 경구 투여된 방사성 동위원소 표지 투여량의 약 4 %였다. 신배설은 투여된 방사성 동위원소 표지량 제거의 부수적 경로이다(2 % 미만).
건강한 성인에서 퀴자티닙의 기하평균(%CV) 전신 청소율(CL)은 2.23 L/hour(29%)로 추정되었다.
(5) 선형성/비 ‑선형성
퀴자티닙은 건강한 성인에서 26.5 mg ~ 79.5 mg 용량 범위에서, 급성 골수성 백혈병 환자에서 17.7 mg ~ 53 mg 용량 범위에서 용량 비례적 선형성을 보였다.
3) 임상시험 정보
이 약의 유효성과 안전성은 FLT3 ‑ITD 변이 양성인 새로 진단받은 급성 골수성 백혈병 18 ‑ 75세 성인 환자(539명)를 대상으로 한 무작위배정, 이중 눈가림, 위약 대조, 3상 임상시험(QuANTUM ‑First)에서 평가되었다. FLT3 ‑ITD 상태는 임상시험 분석법(PCR과 모세관 전기영동법)을 사용하여 전향적으로 확인되었고, FDA에서 승인받은 분석법인 LeukoStratⓡ CDx FLT3 분석 동반진단기기를 사용하여 후향적으로 검증되었다. 환자들은 표준 화학요법과의 병용요법(반응을 보인 환자의 경우 유도요법 후 공고요법) 주기마다 2주간(유도요법 시 8 ‑21일, 공고요법 시 6 ‑19일) 이 약 35.4 mg(n=268) 또는 위약(n=271)을 1일 1회 투여한 후, 이 약(2주간 1일 1회 26.5 mg을 투여하고 이후에는 1일 1회 53 mg을 투여) 또는 위약 단독 유지요법을 최대 36주기(28일/1주기) 동안 투여받도록 무작위배정되었다 (1:1).
환자들은 최대 2주기의 유도 화학요법(다우노루비신 [제1일, 제2일, 제3일에 60 mg/m2/일 IV] 또는 이다루비신 [제1일, 제2일, 제3일에 12 mg/m2/일 IV]과 시타라빈 [7일 동안 100 mg/m2/일 또는 시험기관 또는 현지 표준에 따라 허용되는 경우 200 mg/m2/일, IV 주입]) 후, 최대 4주기의 공고 화학요법 및/또는 HSCT로 구성된 관해 후 치료를 받았다. 공고 화학요법은 60세 미만 환자에 대해 3.0 g/m2 및 60세 이상 환자에 대해 1.5 g/m2의 시타라빈을 제1일, 제3일, 제5일에 12시간마다 IV 주입으로 투여하였다. HSCT를 진행한 환자들은 전처치 시작 7일 전에 시험 치료를 중단하였다.
무작위배정된 두 치료군은 베이스라인 인구통계학적 특성, 질병 특성 및 층화 요인과 관련하여 적절하게 균형을 이루었다. 무작위배정된 539명의 연령 중앙값은 56세(범위 20 ‑75세)였고, 54.5%는 여성, 45.5%는 남성, 59.7%는 백인, 29.3%는 아시아인, 1.3%는 흑인 또는 아프리카계 미국인, 9.7%는 기타 인종이었다. 84%에서 베이스라인 ECOG 전신수행상태(PS) 점수가 0 또는 1이었다. 대다수의 환자(72.4%)가 베이스라인에서 세포유전학적 위험이 중간 상태였다. FLT3 ‑ITD 변이형 대립유전자 빈도(VAF)는 환자의 35.6%에서 3 ‑25%였고, 환자의 52.1%에서는 >25 ‑50%이었고, 환자의 12.1%에서 >50%이었다. 환자의 52%에서 NPM1 변이가 확인되었다.
1차 유효성 평가변수는 무작위 배정일부터 모든 원인에 의한 사망까지의 전체 생존(OS)이었다.
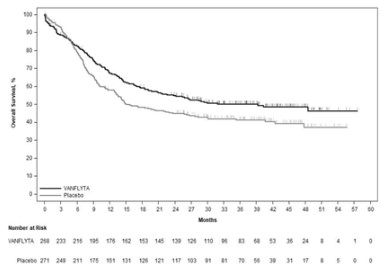
1차 분석은 마지막 환자의 무작위 배정 후 최소 24개월의 추적 관찰 후 수행하였으며 이 약 투여군에서 위약 투여군 대비 전체 생존기간이 통계적으로 유의하게 개선되었다 [위험비(HR) 0.78; 95% CI: 0.62, 0.98; 양측 p=0.0324] (그림 1 참조). 추적관찰 기간의 중앙값은 39.2개월이었다.
이 약과 표준 화학요법과의 병용투여군에서는 유도요법과 공고요법을 각각 65 %, 44 %의 환자가 완료하였고, 위약과 표준 화학요법과의 병용투여군에서는 각각 65 %, 34 %의 환자가 완료하였다. 유지요법을 받은 환자는 208명(39%)이었고, 이 중 64%가 최소 12주기를 완료하였으며, 36%가 최소 24주기, 16%가 36주기를 완료하였다. 유지요법을 받은 208명 중 조혈모세포 이식 없이 공고요법 후 이 약 또는 위약으로 유지 요법을 받은 89명(43%)의 환자를 대상으로 한 탐색적 하위군 분석에서 전체 생존(OS) 위험비(HR)는 0.40 (95% CI: 0.19, 0.84)이었다. 208명 중 조혈모세포 이식 후 이 약 또는 위약으로 유지 요법을 받은 119명(57%)의 환자에서 OS 위험비는 1.62 (95% CI: 0.62, 4.22)이었다.
2차 평가변수인 무사건 생존은 통계적 유의성을 입증하지 못했다.
그림 1: QuANTUM ‑First에서 전체 생존에 대한 카플란 ‑마이어 곡선
4) 독성시험 정보
퀴자티닙을 사용한 발암성 시험은 수행되지 않았다.
유전독성 시험에서, 퀴자티닙은 박테리아 복귀돌연변이 시험에서 변이원성이었으나, 포유류 세포 돌연변이 분석(마우스 림프종 티미딘 키나아제) 또는 생체 내 형질전환 설치류 돌연변이 분석에서는 변이원성이 아니었다. 퀴자티닙은 사람 림프구 염색체 이상 시험 또는 생체 내 랫드 골수 소핵 시험에서 유전독성을 나타내지 않았다.
배 ▪태자 생식 독성 시험에서, 임신한 랫드에게 기관 형성 기간 동안 퀴자티닙을 경구 투여하였다. 6 mg/kg/일 (AUC 기반 53 mg/일인 권장 인체투여량[RHD]의 약 3배)를 투여하였을 때 태자 독성(낮은 태자 체중, 골격 골화에 대한 영향) 및 기형 유발성(부종을 포함한 태자 이상)이 관찰되었다. 10 mg/kg/일 초과의 용량에서, 배 ‑태자 치사성 및 착상 후 손실 증가가 관찰되었다. 모체 독성은 10 mg/kg/일 초과의 용량에서는 체중 증가 감소 및 사료 섭취량 감소로 나타났고, 20 mg/kg/일 용량에서는 이상 임상 징후로 나타났다.
퀴자티닙을 사용한 동물에서의 생식능 시험은 수행되지 않았다. 그러나, 랫드와 원숭이의 반복투여 독성시험에서 수컷 및 암컷 생식기관에 이상적 소견이 관찰되었다. 암컷 랫드의 경우, 10 mg/kg/일(AUC 기반 53 mg/일인 권장 인체투여량 [RHD]의 약 10배)에서 난소 낭종 및 질 점막 변형이 관찰되었다. 암컷 원숭이에서의 소견은 6 mg/kg/일(AUC 기반 RHD의 약 0.3배) 이상의 용량에서 자궁, 난소 및 질 위축이 관찰되었다. 수컷 랫드의 경우, 10 mg/kg/일(AUC 기반 RHD의 약 8배)의 용량에서 정소의 정세관 변성 및 정자 배출 장애가 관찰되었다. 수컷 원숭이에서의 소견은 6 mg/kg/일(AUC 기반 RHD의 약 0.5배) 이상의 용량에서 정소의 생식세포 감소가 관찰되었다. 4주 간의 회복기간 후, 암컷 랫드에서의 질 점막 변형을 제외한 모든 소견은 회복되었다.
FLT3 ‑ITD 변이 양성인 새로 진단받은 급성 골수성 백혈병 성인 환자의 표준 시타라빈 및 안트라사이클린 유도요법과 표준 시타라빈 공고요법과의 병용 및 공고요법 후 단독 유지요법
동종 조혈모세포 이식 후 이 약의 단독 유지요법에 대하여 유효성은 확립되지 않았다.
동종 조혈모세포 이식 후 이 약의 단독 유지요법에 대하여 유효성은 확립되지 않았다.
이 약의 치료 시작 전에 FLT3 ‑ITD 변이 상태를 평가하여야 한다. FLT3 ‑ITD 변이 상태는 충분히 검증된 신뢰성 있는 시험방법을 사용하여 확인하여야 한다. 임상시험시 사용한 진단방법은 '사용상의 주의사항 5. 일반적 주의 1)항 및 14. 전문가를 위한 정보 3)항‘을 참조한다.
1. 용량
이 약은 표준 화학요법과 병용하여 각 유도요법 주기마다 2주간 35.4 mg 용량으로 1일 1회 투여한다. 완전관해(CR) 또는 불완전한 혈액학적 회복을 동반한 완전관해(CRi)에 도달한 환자의 경우, 이 약은 각 공고 화학요법 주기마다 2주간 35.4 mg을 1일 1회 투여한다. 이 약의 유지요법은 공고 화학요법 후 절대 중성구 수 >500/㎣ 및 혈소판 수 >50,000/㎣ 이상으로 회복되면 1일 1회 26.5 mg으로 시작한다. 2주 후, 프리데리시아 공식으로 보정한 QT 간격(QT interval corrected by Fridericia’s formula, QTcF)이 450 ms 이하인 경우, 유지 용량을 1일 1회 53 mg으로 증가한다(표 2 및 ‘3. 신중투여’ 항 참고). 유지 요법은 최대 36주기까지 계속할 수 있다.
추가적인 용량 및 용법에 대한 내용은 표 1 ‑ 3을 참고한다.
표 1: 이 약의 용법용량
조혈모세포 이식(hematopoietic stem cell transplantation, HSCT)이 계획되어 있는 환자의 경우, 이 약은 전처치 7일 전에 중단하여야 한다. 임상시험 중 이 약의 투약에 관한 내용은 ‘14. 전문가를 위한 정보’항을 참고한다.
이 약은 매일 거의 같은 시간에 경구 복용해야 하며 음식과 함께 또는 공복 상태에서 복용할 수 있다.
이 약은 통째로 삼켜야 하며, 씹거나 부수거나 분할해서는 안 된다.
2. 이상반응에 대한 모니터링 및 용량 조절
QTcF가 450 ms 이하인 경우에만 이 약 투여를 시작해야 한다(‘3. 신중투여’항 참고).
유도 및 공고요법 중, 치료 시작 전에 심전도(ECGs) 검사를 수행하고, 이후에는 이 약의 치료 중 주 1회 또는 임상적 증상에 따라 더 자주 수행해야 한다(‘3. 신중투여’항 참고).
유지요법 중, 치료 시작 전에 ECGs 검사를 수행하고, 투여 시작 및 용량 증가 후 처음 1개월 동안에는 주 1회 수행해야 하며, 그 후에는 임상적 증상에 따라 수행해야 한다. QTcF 간격이 450 ms 이하인 경우에만 용량을 증가해야 한다 (표 1 및 ‘3. 신중투여’항 참고).
전해질 이상(저칼륨혈증 및 저마그네슘혈증)은 교정해야 하며, 가능한 경우 QT 간격을 연장하는 약물의 병용투여를 피해야 한다 (‘3. 신중투여’항 참고).
이상반응에 따라 권장되는 용량 조절은 표 2를 참고한다. 이상반응 및/또는 강력한 CYP3A 억제제와 병용투여에 따른 용량 조절은 표 3을 참고한다.
표 2: 이상반응에 따른 용량 조절
3. 이상반응 및/또는 강력한 CYP3A 억제제와 병용투여에 따른 용량 조절
표 3: 이 약으로 치료하는 동안 이상반응 및/또는 강력한 CYP3A 억제제와 병용투여에 따른 단계별 용량 조절
강력한 CYP3A 억제제와 병용투여 시, 퀴자티닙의 노출이 증가할 수 있다. 강력한 CYP3A 억제제와의 병용투여가 불가피한 경우, 표 3에 제시된 대로 이 약의 용량을 감소하도록 한다. 이 약의 용량을 17.7 mg으로 투여하고 있는 경우, 강력한 CYP3A 억제제를 투여하는 동안 이 약의 투여를 중단하고, 강력한 CYP3A 억제제를 중단한 후(반감기의 5배 이상 이후), 이전 용량으로 이 약 투여를 재개한다(‘6. 약물 상호작용’항 참고).
4. 복용 누락 또는 구토
이 약의 복용을 누락하였거나 예정 시간에 복용을 하지 못한 경우, 가능한 빨리 같은 날에 복용하여야 하고, 다음 복용일에 원래의 일정대로 복용하여야 한다. 하루에 2회 용량을 복용하지 않아야 한다.
이 약 복용 후 구토가 있는 경우, 당일 추가 용량을 복용하지 않아야 한다. 다음 복용일에 원래의 일정대로 복용해야 한다.
1. 용량
이 약은 표준 화학요법과 병용하여 각 유도요법 주기마다 2주간 35.4 mg 용량으로 1일 1회 투여한다. 완전관해(CR) 또는 불완전한 혈액학적 회복을 동반한 완전관해(CRi)에 도달한 환자의 경우, 이 약은 각 공고 화학요법 주기마다 2주간 35.4 mg을 1일 1회 투여한다. 이 약의 유지요법은 공고 화학요법 후 절대 중성구 수 >500/㎣ 및 혈소판 수 >50,000/㎣ 이상으로 회복되면 1일 1회 26.5 mg으로 시작한다. 2주 후, 프리데리시아 공식으로 보정한 QT 간격(QT interval corrected by Fridericia’s formula, QTcF)이 450 ms 이하인 경우, 유지 용량을 1일 1회 53 mg으로 증가한다(표 2 및 ‘3. 신중투여’ 항 참고). 유지 요법은 최대 36주기까지 계속할 수 있다.
추가적인 용량 및 용법에 대한 내용은 표 1 ‑ 3을 참고한다.
표 1: 이 약의 용법용량
|
이 약 시작 |
유도a |
공고b |
유지 |
|
제8일에 시작 (7+3 용법)c |
제6일에 시작 |
유지요법 제1일 | |
|
용량 |
1일 1회 35.4mg |
1일 1회 35.4mg |
ㆍ QTcF가 450 ms 이하인 경우 2주간 시작용량으로 1일 1회 26.5 mg 투여 ㆍ 2주 후, QTcF가 450 ms 이하인 경우, 1일 1회 53 mg 투여로 용량을 증가한다. |
|
기간 (28일 주기) |
각 주기마다 2주 |
각 주기마다 2주 |
최대 36주기 동안 주기 간에 휴약기간없이 1일 1회 투여 |
|
a 환자는 최대 2주기의 유도요법을 받을 수 있다. b 환자는 최대 4주기의 공고요법을 받을 수 있다. c 두 번째 유도 주기로서 투여되는 5 + 2 용법의 경우, 이 약은 제6일에 시작한다. | |||
이 약은 매일 거의 같은 시간에 경구 복용해야 하며 음식과 함께 또는 공복 상태에서 복용할 수 있다.
이 약은 통째로 삼켜야 하며, 씹거나 부수거나 분할해서는 안 된다.
2. 이상반응에 대한 모니터링 및 용량 조절
QTcF가 450 ms 이하인 경우에만 이 약 투여를 시작해야 한다(‘3. 신중투여’항 참고).
유도 및 공고요법 중, 치료 시작 전에 심전도(ECGs) 검사를 수행하고, 이후에는 이 약의 치료 중 주 1회 또는 임상적 증상에 따라 더 자주 수행해야 한다(‘3. 신중투여’항 참고).
유지요법 중, 치료 시작 전에 ECGs 검사를 수행하고, 투여 시작 및 용량 증가 후 처음 1개월 동안에는 주 1회 수행해야 하며, 그 후에는 임상적 증상에 따라 수행해야 한다. QTcF 간격이 450 ms 이하인 경우에만 용량을 증가해야 한다 (표 1 및 ‘3. 신중투여’항 참고).
전해질 이상(저칼륨혈증 및 저마그네슘혈증)은 교정해야 하며, 가능한 경우 QT 간격을 연장하는 약물의 병용투여를 피해야 한다 (‘3. 신중투여’항 참고).
이상반응에 따라 권장되는 용량 조절은 표 2를 참고한다. 이상반응 및/또는 강력한 CYP3A 억제제와 병용투여에 따른 용량 조절은 표 3을 참고한다.
표 2: 이상반응에 따른 용량 조절
|
이상반응 |
권장 조치 |
|
QTcF 450 - 480 ms (1등급) |
ㆍ 용량을 유지한다. |
|
QTcF 480 ms 초과 및 500 ms 이하 (2등급) |
ㆍ 투여를 일시 중단하지 않고 용량을 감소한다(표 3 참고). ㆍ QTcF가 450 ms 미만으로 회복되면 다음 주기에서 이전 용량으로 투여를 재개한다. 용량 증가 시 첫 주기 동안 QT 연장에 대해 면밀하게 모니터링한다. |
|
QTcF가 500 ms를 초과한 경우(3등급) |
ㆍ 이 약의 투여를 일시 중단한다. ㆍ QTcF가 450 ms 미만으로 회복되면 용량을 감소하여 투여를 재개한다 (표 3 참고). ㆍ 유도 및/또는 공고요법 중 QTcF의 500 ms 초과가 관찰되고 이 약과 관련 있는 것으로 의심되면, 유지 요법 중 1일 1회 53 mg 투여로 용량을 증가해서는 안 된다. 1일 1회 26.5 mg 용량을 유지한다. |
|
QTcF가 500 ms를 초과한 경우가 재발한 경우(3등급) |
ㆍ 적절한 용량 감소 및 다른 위험 요인(예: 혈청 전해질 이상, QT 간격을 연장시키는 약물 병용)의 교정/제거 조치를 취하였음에도 불구하고, QTcF가 500 ms를 초과한 경우가 재발한 경우, 이 약의 투여를 영구 중단한다. |
|
염전성 심실 빈맥 (Torsades de pointes), 여러 형태 심실 빈맥, 생명을 위협하는 부정맥의 징후/증상(4등급) |
ㆍ 이 약 투여를 영구 중단한다. |
|
3등급 또는 4등급의 비-혈액학적 이상반응 |
ㆍ 이 약의 투여를 일시 중단한다. ㆍ 이상반응이 1등급 이하로 개선되면, 이전 용량으로 투여를 재개한다. ㆍ 이상반응이 3등급 미만으로 개선되면, 용량을 감소하여 투여를 재개한다(표 3 참고). ㆍ 3등급 또는 4등급 이상반응이 28일 이상 지속되고 이 약과 관련 있는 것으로 의심되는 경우, 투여를 중단한다. |
|
활동성 골수 질환이 없는 지속성 4등급 중성구 감소증 또는 혈소판 감소증 |
ㆍ 용량을 감소한다(표 3 참고). |
|
이상반응은 National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03(NCI CTCAE v4.03)에 따른다. | |
표 3: 이 약으로 치료하는 동안 이상반응 및/또는 강력한 CYP3A 억제제와 병용투여에 따른 단계별 용량 조절
|
치료 단계 |
전체 용량 |
용량 감소 | ||
|
이상반응 발생 |
강력한 CYP3A 억제제와 병용투여 |
이상반응 발생 및 강력한 CYP3A 억제제와 병용투여 | ||
|
유도 또는 공고 |
35.4 mg |
26.5 mg |
17.7 mg |
투여 일시 중단 |
|
유지 (처음 2주) |
26.5 mg |
투여 일시 중단 |
17.7 mg |
투여 일시 중단 |
|
유지 (2주 후) |
53 mg |
35.4 mg |
26.5 mg |
17.7 mg |
4. 복용 누락 또는 구토
이 약의 복용을 누락하였거나 예정 시간에 복용을 하지 못한 경우, 가능한 빨리 같은 날에 복용하여야 하고, 다음 복용일에 원래의 일정대로 복용하여야 한다. 하루에 2회 용량을 복용하지 않아야 한다.
이 약 복용 후 구토가 있는 경우, 당일 추가 용량을 복용하지 않아야 한다. 다음 복용일에 원래의 일정대로 복용해야 한다.
1. 경고 (‘3. 신중투여’항 참고)
1) QT 간격 연장
이 약으로 치료받은 환자에서 염전성 심실 빈맥(torsades de pointes), 심실세동, 심정지 및 급사가 발생했다.
임상시험에서 이 약으로 치료받은 급성 골수성 백혈병 환자 1,081명 중 약 0.1 %에서 염전성 심실 빈맥(torsades de pointes)이 발생했고, 0.6 %에서 심정지가 발생했으며 그 중 0.4 %는 치명적인 결과를 나타냈고, 0.1 %의 환자에서 심실세동이 발생했다. 이러한 중증 부정맥은 주로 유도 단계에서 발생했다.
2. 다음 환자에는 투여하지 말 것
1) 이 약의 주성분 또는 구성성분에 과민증이 있는 환자
2) QT 연장 증후군, 심실성 부정맥 또는 염전성 심실 빈맥(torsades de pointes) 병력, 중증 저칼륨혈증, 중증 저마그네슘혈증이 있는 환자(‘3. 신중투여’항 참고).
3. 다음 환자에는 신중히 투여할 것
1) QT 간격 연장
이 약은 QT 간격 연장과 관련이 있다. QT 간격 연장은 심실성 부정맥 또는 염전성 심실 빈맥(torsades de pointes)의 위험성을 증가시킬 수 있다. 선천성 QT 연장 증후군 및/또는 염전성 심실 빈맥(torsades de pointes)의 병력이 있는 환자는 퀴자티닙의 개발 프로그램에서 제외되었다. QT 연장 증후군, 심실성 부정맥 또는 염전성 심실 빈맥(torsades de pointes) 병력, 중증 저칼륨혈증, 중증 저마그네슘혈증이 있는 환자에게 이 약을 투여해서는 안 된다.
이 약은 QT 간격 연장이 발생할 유의한 위험이 있는 환자에게 주의해서 사용해야 한다. 이는 조절되지 않거나 유의한 심혈관 질환(예. 2도 또는 3도 심장차단(박동조율기 없이), 6개월 이내 심근경색, 조절되지 않는 협심증, 조절되지 않는 고혈압), 울혈성 심부전 병력이 있는 환자 및 QT 간격을 연장시키는 것으로 알려진 약물을 병용 투여한 환자를 포함한다. 전해질은 정상 범위로 유지되어야 한다 (‘용법용량’ 참조).
이 약의 치료 시작 전 ECGs 검사를 수행해야 하고, 전해질 이상이 있는 경우 투여 시작 전 교정하여야 한다. QTcF 간격이 450 ms 초과인 경우, 이 약 투여를 시작해서는 안 된다.
유도 및 공고요법 중, ECGs 검사는 투여 시작 전 수행하고, 이후에는 이 약 투여 중 주 1회 또는 임상적 증상에 따라 더 자주 수행해야 한다.
유지요법 중, ECGs 검사는 투여 시작 전 수행하고, 투여 시작 및 용량 증가 후 처음 1개월 동안에는 주 1회 수행해야 하며, 그 후에는 임상적 증상에 따라 수행해야 한다. QTcF 간격이 450 ms 초과인 경우 유지 시작 용량을 증가해서는 안 된다(표 1 참고).
생명을 위협하는 부정맥의 징후 또는 증상과 함께 QT 간격 연장이 나타난 환자에 대해서는 이 약을 영구 중단한다(‘용법 ▪용량’ 참고).
QT 간격의 ECG 모니터링은 QT 간격 연장 및 염전성 심실 빈맥(torsades de pointes)이 발생할 위험이 유의하게 있는 환자에서 더 자주 수행되어야 한다.
이 약의 치료 시작 전 및 치료 중에 저칼륨혈증 및 저마그네슘혈증의 모니터링 및 교정을 수행해야 한다. 설사나 구토를 경험한 환자에서는 전해질 및 심전도를 더 자주 모니터링 해야 한다.
⓵ QT 간격을 연장하는 약물 투여 시 ECG 모니터링
이 약과 QT 간격을 연장하는 것으로 알려진 약물을 함께 투여하는 경우, 환자의 ECG를 더 자주 모니터링 해야 한다. QT 간격을 연장하는 약물의 예는 아졸계 항진균제, 온단세트론, 그라니세트론, 아지스로마이신, 펜타미딘, 독시사이클린, 목시플록사신, 아토바쿠온, 프로클로르페라진 및 타크로리무스가 포함되지만 이에 국한되지 않는다.
⓶ 강력한 CYP3A 억제제와 병용 투여
이 약과 강력한 CYP3A 억제제를 병용 투여하는 경우, 퀴자티닙의 노출을 증가시킬 수도 있으므로 이 약의 용량을 감소해야 한다 (‘용법 ▪용량’ 참고).
2) 배 ▪태아 독성
동물 시험 결과에 근거하여, 이 약은 임부에게 투여 시 배 ▪태아에 위해할 수 있다. 임부에게 태아에 대한 잠재적 위험에 대해 알린다. 임신 가능성이 있는 여성에게 이 약 투여 중 및 마지막 투여 이후 최소 7개월 동안 효과적인 피임법을 사용해야 한다. 임신 가능성이 있는 배우자가 있는 남성은 이 약 투여 중 및 마지막 투여 이후 최소 4개월 동안 효과적인 피임법을 사용해야 한다.(‘7. 임부 및 수유부에 대한 투여’항 참고).
4. 이상반응
1) 안전성 프로파일 요약
이 약의 안전성은 FLT3 ‑ITD 양성인 새로 진단받은 급성 골수성 백혈병 성인 환자를 대상으로 한 무작위배정, 이중 눈가림, 위약 대조 임상시험(QuANTUM ‑First)에서 조사되었다. 환자들은 이 약(N=265) 또는 위약(N=268)을 표준 화학요법과 병용 투여받거나 단독 유지요법으로 투여받았다(‘14. 전문가를 위한 정보’항 참고). 전체 치료 기간의 중앙값은 이 약 치료군의 환자에 대해서는 10.7주(범위: 0.1 ~ 184.1)였고 위약 치료군의 환자에 대해서는 9.5주(범위: 0.4 ~ 181.9)였다.
전체 시험기간 중, 이 약 치료군에서 가장 흔하게 보고된 이상반응(발생률 >20%)은 알라닌 아미노 전이 효소 증가(58.9%), 혈소판 수 감소(40.0%), 헤모글로빈 감소(37.4%), 설사(37.0%), 구역(34.0%), 복통(29.4%), 두통(27.5%), 구토(24.5%) 및 중성구 수 감소(21.9%)였다. 가장 흔하게 보고된 3등급 또는 4등급 이상반응(발생률 ≧5%)은 혈소판 수 감소(40.0%), 헤모글로빈 감소(35.5%), 중성구 수 감소(21.5%), 알라닌 아미노 전이 효소 증가(12.1%) 균혈증(7.2%) 및 진균 감염(5.7%)이었다.
이 약 치료군에서 가장 흔하게 보고된 중대한 이상반응(발생률 ≧2%)은 중성구 감소증(3.0%), 진균 감염(2.3%) 및 헤르페스 감염(2.3%)이었다. 치명적인 결과를 나타낸 이상반응은 진균 감염(0.8%) 및 심정지(0.4%)였다.
이 약의 투여 일시 중단과 연관된 가장 흔하게 보고된 이상반응(발생률 ≧2%)은 중성구 감소증(10.6%), 혈소판 감소증(4.5%) 및 심전도 QT 연장(2.6%)이었다. 이 약의 용량 감소와 연관된 가장 흔하게 보고된 이상반응(발생률 ≧2%)은 중성구 감소증(9.1%), 혈소판 감소증(4.5%) 및 심전도 QT 연장(3.8%)이었다.
이 약의 영구 중단과 연관된 가장 흔하게 보고된 이상반응(발생률 ≧1%)은 혈소판 감소증(1.1%)이었다.
단독 유지요법 중 안전성 프로파일
유지 단계에 진입한 208명(39%)의 환자는 이 약(116명) 또는 위약(92명)을 투여받았다. 유지 요법을 위한 치료기간의 중앙값은 이 약 치료군의 환자에 대해서는 67.4주(범위: 0.4 ~ 167.0)였고 위약 치료군의 환자에 대해서는 67.7주(범위: 0.3 ~ 167.4)였다.
이 약 치료군에서 가장 흔하게 보고된 이상반응은 중성구 수 감소(87.9%), 혈소판 수 감소(72.4%), 헤모글로빈 감소(55.2%), 알라닌 아미노 전이 효소 증가(49.1%), 상기도 감염(27.6%), 구역(23.3%) 및 설사(20.7%)였다. 이 약 투여 시 가장 빈번하게 보고된 3등급 또는 4등급 이상반응은 중성구 수 감소(71.6%), 혈소판 수 감소(24.1%) 및 헤모글로빈 감소(18.1%)였다.
이 약 치료군에서 가장 흔하게 보고된 중대한 이상반응은 헤르페스 감염(3.4%), 중성구 감소증(2.6%), 상기도 감염(1.7%) 및 구토(1.7%)였다. 유지 요법 중 치명적인 이상반응은 발생하지 않았다.
이 약의 투여 일시 중단과 연관된 가장 흔하게 보고된 이상반응은 중성구 감소증(24.1%), 혈소판 감소증(10.3%) 및 심전도 QT 연장(4.3%)이었다. 이 약의 용량 감소와 연관된 가장 흔하게 보고된 이상반응은 중성구 감소증(20.7%), 혈소판 감소증(10.3%) 및 심전도 QT 연장(5.2%)이었다. 이 약의 영구 중단과 연관된 가장 흔하게 보고된 이상반응은 혈소판 감소증(2.6%), 구역, 중성구 감소증 및 식욕 감소(각각 1.7%)였다.
2) 이상반응 표
이상반응은 MedDRA 기관계 대분류(SOC)에 따라 기재되었다. 각 기관계 내에서, 이상반응은 다음의 통상의 규칙(CIOMS III)을 이용하여 가장 빈도가 높은 반응 순으로 빈도별로 정렬하였다: 매우 흔하게( ≧1/10), 흔하게( ≧1/100 ~ <1/10), 흔하지 않게( ≧1/1000 ~ <1/100), 드물게( ≧1/10,000 ~ <1/1000), 매우 드물게(<1/10,000). 각 빈도 범주 내에서 이상반응은 중증도가 감소하는 순서대로 기재하였다.
표 4: 이 약의 임상시험(QuANTUM ‑First)에서 관찰된 이상반응
3) 심장 장애
이 약은 ECG에서 QT 간격을 연장한다. 등급에 관계없이, 치료 후 발생한 이상반응으로써 QT 간격 연장은 이 약을 투여한 환자의 14.0%에서 보고되었고, 환자의 3.0%가 3등급 이상의 이상반응을 경험하였다. QT 연장은 환자 10명(3.8%)에서는 용량 감소, 7명(2.6%)에서는 투여 일시 중단, 2명(0.8%)에서는 영구 중단을 초래하였다. 중앙에서 검토된 ECG 데이터에 근거하여, QTcF >500 ms는 환자의 2.3%에서 발생하였다. 이 약을 투여한 환자 2명(0.8%)이 심실세동 기록과 함께 심정지를 경험하였고, 1명은 치명적인 결과를 나타냈으며, 이들 모두 중증 저칼륨혈증이 있는 상황에서 발생하였다. 이 약 투여 전 및 투여 중 심전도 검사 및 모니터링, 저칼륨혈증 및 저마그네슘혈증 교정을 실시해야 한다. QT 간격 연장이 있는 환자에 대한 이 약의 용량 조절은 ‘용법 ▪용량’ 항을 참고한다.
5. 일반적 주의
1) FLT3 ‑ITD 변이 검사
이 약은 FLT3 ‑ITD 변이 양성인 급성 골수성 백혈병 환자에서만 치료 이익을 보였으므로, 이 약의 치료를 받을 환자를 선택하기 위해서는 FLT3 ‑ITD 변이 양성인 급성 골수성 백혈병의 진단이 필요하다. FLT3 ‑ITD 상태는 임상시험 분석법(PCR과 모세관 전기영동법)을 사용하여 전향적으로 확인되었고, FDA에서 승인받은 분석법인 LeukoStratⓡ CDx FLT3 분석 동반진단기기를 사용하여 후향적으로 검증되었다.
6. 약물 상호작용
1) 강력한 CYP3A 억제제
건강한 대상자에게 이 약과 강력한 CYP3A 억제제와 병용투여 시, 이 약 단독 투여와 비교하여 퀴자티닙의 노출이 증가하였다. 이 약의 1회 투여 용량(26.5 mg)과 케토코나졸(200 mg씩 1일 2회)을 병용투여 시 퀴자티닙의 Cmax는 17 %, AUCinf는 94 % 증가하였다. 활성 대사체인 AC886의 AUCinf는 15 %, Cmax는 60 % 감소했다. 정상 상태에서 퀴자티닙의 노출(Cmax 및 AUC0 ‑24h)은 각각 86 %, 96 % 증가할 것으로 추정되었다. 퀴자티닙의 노출 증가로 독성의 위험성이 증가할 수도 있다.
강력한 CYP3A 억제제와의 병용투여가 불가피한 경우, 이 약의 용량을 감소해야 한다(‘용법용량’ 참고). 강력한 CYP3A 억제제의 예로 케토코나졸, 이트라코나졸, 포사코나졸, 보리코나졸, 클래리스로마이신, 네파조돈, 텔리스로마이신 및 항레트로바이러스제가 있다.
2) 강력한 및 중등도의 CYP3A 유도제
건강한 대상자에게 이 약과 중등도의 CYP3A 유도제의 병용투여 시, 이 약 단독 투여(총 53 mg)와 비교하여 퀴자티닙 및 활성 대사체 AC886의 노출이 감소하였다. 중등도의 CYP3A 유도제인 에파비렌즈(600 mg 씩 1일 1회)와 병용투여 시 퀴자티닙 Cmax 및 AUCinf는 각각 약 45 %와 90 % 감소하였다. AC886의 Cmax 및 AUCinf는 각각 약 68 % 및 96 % 감소하였다 (‘14. 전문가용 정보’항 참고).
퀴자티닙 노출 감소는 유효성 감소로 이어질 수 있다. 이 약과 강력한 또는 중등도의 CYP3A 유도제와의 병용투여를 피해야 한다.
강력한 CYP3A4 유도제의 예로는 아팔루타마이드, 카르바마제핀, 엔잘루타마이드, 미토탄, 페니토인, 리팜피신, 세인트 존스 워트(Hypericum perforatum 라고도 함) 등 특정 생약 제제가 있다. 중등도의 CYP3A4 유도제의 예로는 에파비렌즈, 보젠탄, 에트라비린, 페노바르비탈 및 프리미돈이 있다.
3) 수송체
⓵ P ‑당단백(P ‑gp) 기질
퀴자티닙과 다비가트란 에텍실레이트(P ‑gp 기질)를 병용투여하였을 때, 총 및 유리 다비가트란의 Cmax는 각각 12% 및 13% 증가하였고, AUCinf는 각각 13% 및 11% 증가하였다(‘14. 전문가용 정보’항 참고). 퀴자티닙은 약한 P ‑gp 억제제이며, 이 약과 P ‑gp 기질을 병용투여 시 용량 조절은 권장되지 않는다.
⓶ 유방암 내성 단백질(BCRP) 기질
퀴자티닙은 in vitro IC50 추정값 0.813 μM에서 유방암 내성 단백질을 저해한다. 생리학적 기반 약동학(PBPK) 분석에 따르면 퀴자티닙 53mg은 로수바스타틴의 AUC0 ‑48h를 2.44배, 설파살라진은 2.66배 증가시킬 것으로 예측되었다. 퀴자티닙을 BCRP 기질 약물과 병용투여할 경우 주의해야 한다. BCRP 기질 약물의 용량 감소를 고려해야 한다.
⓷ Uridine Diphosphate Glucuronosyltransferases(UGT) 1A1 기질
퀴자티닙은 in vitro Ki 추정값 0.78 μM에서 UGT1A1을 저해한다. PBPK 분석에 근거하여, 퀴자티닙은 랄테그라비르(UGT1A1 기질)의 Cmax와 AUCinf를 3% 증가시킬 것으로 예상되었다. UGT1A1 기질과 이 약의 병용투여 시 용량 조절은 권장되지 않는다.
⓸ QT 간격을 연장시키는 약물
이 약과 QT 간격을 연장하는 기타 약물의 병용투여는 QT 연장의 발생률을 더욱 증가시킬 수 있다. 이 약과 QT 간격을 연장하는 약물을 병용투여 시 주의해야 한다(‘3. 다음 환자에는 신중히 투여할 것 중 ‘1) QT 간격 연장’항 참고).
7. 임부 및 수유부에 대한 투여
1) 남성 및 여성의 피임
이 약은 임부에게 투여 시 배 ▪태아에 해를 끼칠 수 있다(‘14. 전문가용 정보’항 참고). 잠재적인 임신 가능성이 있는 여성은 이 약 투여 중 및 마지막 투여 후 최소 7개월 동안 효과적인 피임법을 사용하여야 한다. 임신 가능성이 있는 여성 배우자가 있는 남성은 이 약 투여 중 및 마지막 투여 후 최소 4개월 동안 효과적인 피임법을 사용하여야 한다.
2) 임부
임부에게 퀴자티닙을 투여한 자료는 없다. 동물시험 결과에 근거하여, 이 약은 임부에게 투여 시 배 ▪태아 독성을 초래할 수 있다 (‘14. 전문가용 정보’항 참고).
이 약을 임신 중 투여해서는 안 된다. 임부에게 태아에 대한 잠재적 위험에 대해 알린다.
3) 수유부
퀴자티닙 또는 활성 대사체가 모유로 분비되는지 여부는 알려져 있지 않다. 모유 수유 중인 소아에서 중대한 이상반응이 발생할 가능성이 있기 때문에, 이 약 투여 중 및 마지막 투여 후 최소 5주 동안 모유 수유를 해서는 안 된다.
4) 임신 가능성이 있는 여성
임신 가능성이 있는 여성은 이 약으로 치료를 시작하기 전 임신 검사를 받아야 한다.
5) 생식능
이 약이 사람의 생식능에 미치는 영향에 대한 자료는 없다. 동물 시험 결과에 근거할 때, 이 약 투여로 인해 여성 및 남성 생식능이 손상될 수 있다(‘14. 전문가를 위한 정보’항 참고).
8. 소아에 대한 투여
이 약은 18세 미만의 소아 및 청소년에서 안전성 및 유효성이 확립되지 않았다. 가용할 수 있는 데이터가 없다.
9. 고령자에 대한 투여
65세 이상 환자에서 이 약의 용량 조절은 필요하지 않다.
임상시험(QuANTUM ‑First)에서 이 약을 투여받은 새로 진단받은 급성 골수성 백혈병 환자 265명 중 69명(26%)이 65세 이상이었고 1명(0.4%)이 75세였다. 65세 이상 환자와 젊은 성인 환자 간에 안전성에서 전반적인 차이는 관찰되지 않았다.
이 약의 임상시험에는 젊은 성인 환자와 다르게 반응을 보이는지를 확인하기에 충분한 수의 65세 이상 환자가 포함되지 않았다.
이 약은 특히 치료 초기 단계에서 젊은 환자에 비해 고령 환자(즉, 65세 이상)에서 치명적인 감염 발생 빈도가 더 높았다. 65세 이상 환자는 유도 요법 기간 동안 중증 감염 발생 여부를 면밀히 모니터링해야 한다.
10. 신 장애 환자에 대한 투여
경증 또는 중등도 신 장애 환자에서 용량 조절은 권장되지 않는다.
이 약은 중증 신 장애(Cockcroft ‑Gault 식으로 추정한 CLcr <30 mL/min) 환자에서 안전성 및 유효성이 평가되지 않았기 때문에, 중증 신 장애 환자에게는 권장되지 않는다.
11. 간 장애 환자에 대한 투여
경증 또는 중등도 간 장애 환자에서 용량 조절은 권장되지 않는다.
이 약은 중증 간 장애(Child ‑Pugh Class C 또는 총 빌리루빈 >3 x ULN 및 모든 수치의 AST) 환자에서 안전성 및 유효성이 평가되지 않았기 때문에, 중증 간 장애 환자에게는 권장되지 않는다.
12. 과량투여시의 처치
이 약의 과량투여에 대한 해독제는 알려지지 않았다. 과량투여 시 투여를 일시 중단하고, QT 간격 연장을 측정하기 위한 ECG를 수행하며, 적절한 보조 조치를 한다(‘용법용량’ 및 ‘3. 신중투여’항 참고).
13. 보관 및 취급 시 주의사항
1) 이 약은 어린의 손이 닿지 않는 곳에 보관해야 한다.
2) 이 약을 다른 용기에 옮겨 담지 않는다.
14. 전문가를 위한 정보
1) 약리작용
(1) 작용 기전
퀴자티닙은 티로신 키나아제 FLT3 수용체의 저분자 억제제다. 퀴자티닙 및 그 주요 순환 활성 대사체 AC886은 FLT3의 아데노신삼인산(ATP) 결합 부위에 높은 친화성(각각 Kd = 1.3 nM 및 0.54 nM)으로 경쟁적으로 결합한다. 퀴자티닙 및 AC886은 수용체의 자가인산화를 방지하여 FLT3 키나아제 활성을 억제하며, 이후 다운스트림 FLT3 수용체 신호전달을 억제하고 FLT3 ‑ITD 의존적인 세포 증식을 차단한다.
(2) 약력학적 효과
심장 전기생리학
임상시험(QuANTUM ‑First)의 노출 ‑반응 분석에서 유지요법 중 퀴자티닙(53 mg)의 정상 상태 Cmax에서 연장된 QTcF 간격은 24.1 ms(양측 90% CI의 상한: 26.6 ms)였으며 농도 ‑의존적인 QTcF 간격 연장이 예상되었다.
2) 약동학적 특성
퀴자티닙 및 주요 순환 활성 대사체(AC886)의 약동학은 건강한 성인과 성인 암 환자에서 분석되었으며, 달리 명시되지 않는 한 기하 평균(변동계수 [%CV])으로 제시되었다.
급성 골수성 백혈병 성인 환자에서 1일 1회 투여 후 15일 째에 퀴자티닙 농도가 정상 상태(steady state)에 도달했다. 건강한 성인의 경우 퀴자티닙 노출(AUC 및 Cmax)은 26.5 mg (권장 유도 용량 35.4 mg의 0.75배)에서 79.5 mg (권장 유도 용량 35.4 mg의 2.25배)까지, 급성 골수성 백혈병 성인 환자의 경우 17.7mg (권장 유도 용량 35.4mg의 0.5배)에서 53 mg (권장 유도 용량 35.4 mg의 1.5배)까지의 용량 범위에서 비례적으로 증가했다.
표 5. 퀴자티닙 및 AC886의 약동학적 특성
*평균(±표준편차)(Mean (±SD))
(1) 흡수
정제 제형에서 퀴자티닙의 절대 생체이용률은 71 %였다. 건강한 성인에게 공복 상태에서 경구 투여 시 퀴자티닙 및 AC886의 최고 농도 도달 시간(Tmax 중앙값)은 각각 약 4시간(범위 2 ‑ 8시간) 및 5 ‑ 6시간(범위 4 ‑ 120시간)이었다. 53 mg/일 용량으로 유지요법 중 정상 상태에서, 퀴자티닙 및 AC886의 기하평균(%CV) Cmax는 각각 529 ng/mL(60 %) 및 262 ng/mL(48 %)로 추정되었고, 기하평균(%CV) AUC0 ‑24h는 각각 10,200 ng ▪h/mL(75 %) 및 5,790 ng ▪h/mL(46 %)였다.
음식과 함께 이 약 투여 시 퀴자티닙 Cmax는 8% 감소하였으며, AUCinf는 8% 증가하였다. Tmax는 2시간 지연되었다. 이 약은 음식 여부와 관계없이 투여 가능하다.
(2) 분포
퀴자티닙 및 AC886의 In vitro 사람 혈장 단백질 결합률은 각각 99% 이상이다.
10 ‑ 1,000 ng/mL 농도 범위에서 in vitro 적혈구 내 퀴자티닙 및 AC886의 혈액 대 혈장 비율은 각각 0.79 ‑ 1.30 및 1.36 ‑ 3.19이다.
건강한 성인에서 퀴자티닙의 정상 상태에서 분포 용적(Vss)의 기하평균(%CV)은 275 L(17%)로 추정되었다.
(3) 생체 내 변환
퀴자티닙은 활성 대사체 AC886을 생성하는 산화 경로를 통해 in vitro에서 CYP3A4/5에 의해 주로 대사되며, 이후 CYP3A4/5에 의해 추가로 대사된다. 유지요법 중 정상 상태에서 퀴자티닙에 대한 AC886의 AUC0 ‑24h비는 0.57이었다.
(4) 배설
퀴자티닙 및 AC886의 유효 반감기(t1/2)의 평균(표준편차)는 새로 진단된 급성 골수성 백혈병 환자에서 각각 81시간(±73) 및 136시간(±113)이다. 퀴자티닙 및 AC886의 축적비의 평균 AUC0 ‑24h(표준편차)는 각각 5.4(±4.4) 및 8.7(±6.8)이었다.
퀴자티닙 및 그 대사체는 대부분이 일차적으로 간담도 경로를 통해 제거되어 대변으로 배설된다(경구 투여된 방사성 동위원소 표지 투여량의 76.3 %). 대변으로 배설되는 미변화체 퀴자티닙은 경구 투여된 방사성 동위원소 표지 투여량의 약 4 %였다. 신배설은 투여된 방사성 동위원소 표지량 제거의 부수적 경로이다(2 % 미만).
건강한 성인에서 퀴자티닙의 기하평균(%CV) 전신 청소율(CL)은 2.23 L/hour(29%)로 추정되었다.
(5) 선형성/비 ‑선형성
퀴자티닙은 건강한 성인에서 26.5 mg ~ 79.5 mg 용량 범위에서, 급성 골수성 백혈병 환자에서 17.7 mg ~ 53 mg 용량 범위에서 용량 비례적 선형성을 보였다.
3) 임상시험 정보
이 약의 유효성과 안전성은 FLT3 ‑ITD 변이 양성인 새로 진단받은 급성 골수성 백혈병 18 ‑ 75세 성인 환자(539명)를 대상으로 한 무작위배정, 이중 눈가림, 위약 대조, 3상 임상시험(QuANTUM ‑First)에서 평가되었다. FLT3 ‑ITD 상태는 임상시험 분석법(PCR과 모세관 전기영동법)을 사용하여 전향적으로 확인되었고, FDA에서 승인받은 분석법인 LeukoStratⓡ CDx FLT3 분석 동반진단기기를 사용하여 후향적으로 검증되었다. 환자들은 표준 화학요법과의 병용요법(반응을 보인 환자의 경우 유도요법 후 공고요법) 주기마다 2주간(유도요법 시 8 ‑21일, 공고요법 시 6 ‑19일) 이 약 35.4 mg(n=268) 또는 위약(n=271)을 1일 1회 투여한 후, 이 약(2주간 1일 1회 26.5 mg을 투여하고 이후에는 1일 1회 53 mg을 투여) 또는 위약 단독 유지요법을 최대 36주기(28일/1주기) 동안 투여받도록 무작위배정되었다 (1:1).
환자들은 최대 2주기의 유도 화학요법(다우노루비신 [제1일, 제2일, 제3일에 60 mg/m2/일 IV] 또는 이다루비신 [제1일, 제2일, 제3일에 12 mg/m2/일 IV]과 시타라빈 [7일 동안 100 mg/m2/일 또는 시험기관 또는 현지 표준에 따라 허용되는 경우 200 mg/m2/일, IV 주입]) 후, 최대 4주기의 공고 화학요법 및/또는 HSCT로 구성된 관해 후 치료를 받았다. 공고 화학요법은 60세 미만 환자에 대해 3.0 g/m2 및 60세 이상 환자에 대해 1.5 g/m2의 시타라빈을 제1일, 제3일, 제5일에 12시간마다 IV 주입으로 투여하였다. HSCT를 진행한 환자들은 전처치 시작 7일 전에 시험 치료를 중단하였다.
무작위배정된 두 치료군은 베이스라인 인구통계학적 특성, 질병 특성 및 층화 요인과 관련하여 적절하게 균형을 이루었다. 무작위배정된 539명의 연령 중앙값은 56세(범위 20 ‑75세)였고, 54.5%는 여성, 45.5%는 남성, 59.7%는 백인, 29.3%는 아시아인, 1.3%는 흑인 또는 아프리카계 미국인, 9.7%는 기타 인종이었다. 84%에서 베이스라인 ECOG 전신수행상태(PS) 점수가 0 또는 1이었다. 대다수의 환자(72.4%)가 베이스라인에서 세포유전학적 위험이 중간 상태였다. FLT3 ‑ITD 변이형 대립유전자 빈도(VAF)는 환자의 35.6%에서 3 ‑25%였고, 환자의 52.1%에서는 >25 ‑50%이었고, 환자의 12.1%에서 >50%이었다. 환자의 52%에서 NPM1 변이가 확인되었다.
1차 유효성 평가변수는 무작위 배정일부터 모든 원인에 의한 사망까지의 전체 생존(OS)이었다.
1차 분석은 마지막 환자의 무작위 배정 후 최소 24개월의 추적 관찰 후 수행하였으며 이 약 투여군에서 위약 투여군 대비 전체 생존기간이 통계적으로 유의하게 개선되었다 [위험비(HR) 0.78; 95% CI: 0.62, 0.98; 양측 p=0.0324] (그림 1 참조). 추적관찰 기간의 중앙값은 39.2개월이었다.
이 약과 표준 화학요법과의 병용투여군에서는 유도요법과 공고요법을 각각 65 %, 44 %의 환자가 완료하였고, 위약과 표준 화학요법과의 병용투여군에서는 각각 65 %, 34 %의 환자가 완료하였다. 유지요법을 받은 환자는 208명(39%)이었고, 이 중 64%가 최소 12주기를 완료하였으며, 36%가 최소 24주기, 16%가 36주기를 완료하였다. 유지요법을 받은 208명 중 조혈모세포 이식 없이 공고요법 후 이 약 또는 위약으로 유지 요법을 받은 89명(43%)의 환자를 대상으로 한 탐색적 하위군 분석에서 전체 생존(OS) 위험비(HR)는 0.40 (95% CI: 0.19, 0.84)이었다. 208명 중 조혈모세포 이식 후 이 약 또는 위약으로 유지 요법을 받은 119명(57%)의 환자에서 OS 위험비는 1.62 (95% CI: 0.62, 4.22)이었다.
2차 평가변수인 무사건 생존은 통계적 유의성을 입증하지 못했다.
그림 1: QuANTUM ‑First에서 전체 생존에 대한 카플란 ‑마이어 곡선
4) 독성시험 정보
퀴자티닙을 사용한 발암성 시험은 수행되지 않았다.
유전독성 시험에서, 퀴자티닙은 박테리아 복귀돌연변이 시험에서 변이원성이었으나, 포유류 세포 돌연변이 분석(마우스 림프종 티미딘 키나아제) 또는 생체 내 형질전환 설치류 돌연변이 분석에서는 변이원성이 아니었다. 퀴자티닙은 사람 림프구 염색체 이상 시험 또는 생체 내 랫드 골수 소핵 시험에서 유전독성을 나타내지 않았다.
배 ▪태자 생식 독성 시험에서, 임신한 랫드에게 기관 형성 기간 동안 퀴자티닙을 경구 투여하였다. 6 mg/kg/일 (AUC 기반 53 mg/일인 권장 인체투여량[RHD]의 약 3배)를 투여하였을 때 태자 독성(낮은 태자 체중, 골격 골화에 대한 영향) 및 기형 유발성(부종을 포함한 태자 이상)이 관찰되었다. 10 mg/kg/일 초과의 용량에서, 배 ‑태자 치사성 및 착상 후 손실 증가가 관찰되었다. 모체 독성은 10 mg/kg/일 초과의 용량에서는 체중 증가 감소 및 사료 섭취량 감소로 나타났고, 20 mg/kg/일 용량에서는 이상 임상 징후로 나타났다.
퀴자티닙을 사용한 동물에서의 생식능 시험은 수행되지 않았다. 그러나, 랫드와 원숭이의 반복투여 독성시험에서 수컷 및 암컷 생식기관에 이상적 소견이 관찰되었다. 암컷 랫드의 경우, 10 mg/kg/일(AUC 기반 53 mg/일인 권장 인체투여량 [RHD]의 약 10배)에서 난소 낭종 및 질 점막 변형이 관찰되었다. 암컷 원숭이에서의 소견은 6 mg/kg/일(AUC 기반 RHD의 약 0.3배) 이상의 용량에서 자궁, 난소 및 질 위축이 관찰되었다. 수컷 랫드의 경우, 10 mg/kg/일(AUC 기반 RHD의 약 8배)의 용량에서 정소의 정세관 변성 및 정자 배출 장애가 관찰되었다. 수컷 원숭이에서의 소견은 6 mg/kg/일(AUC 기반 RHD의 약 0.5배) 이상의 용량에서 정소의 생식세포 감소가 관찰되었다. 4주 간의 회복기간 후, 암컷 랫드에서의 질 점막 변형을 제외한 모든 소견은 회복되었다.
1) QT 간격 연장
이 약으로 치료받은 환자에서 염전성 심실 빈맥(torsades de pointes), 심실세동, 심정지 및 급사가 발생했다.
임상시험에서 이 약으로 치료받은 급성 골수성 백혈병 환자 1,081명 중 약 0.1 %에서 염전성 심실 빈맥(torsades de pointes)이 발생했고, 0.6 %에서 심정지가 발생했으며 그 중 0.4 %는 치명적인 결과를 나타냈고, 0.1 %의 환자에서 심실세동이 발생했다. 이러한 중증 부정맥은 주로 유도 단계에서 발생했다.
2. 다음 환자에는 투여하지 말 것
1) 이 약의 주성분 또는 구성성분에 과민증이 있는 환자
2) QT 연장 증후군, 심실성 부정맥 또는 염전성 심실 빈맥(torsades de pointes) 병력, 중증 저칼륨혈증, 중증 저마그네슘혈증이 있는 환자(‘3. 신중투여’항 참고).
3. 다음 환자에는 신중히 투여할 것
1) QT 간격 연장
이 약은 QT 간격 연장과 관련이 있다. QT 간격 연장은 심실성 부정맥 또는 염전성 심실 빈맥(torsades de pointes)의 위험성을 증가시킬 수 있다. 선천성 QT 연장 증후군 및/또는 염전성 심실 빈맥(torsades de pointes)의 병력이 있는 환자는 퀴자티닙의 개발 프로그램에서 제외되었다. QT 연장 증후군, 심실성 부정맥 또는 염전성 심실 빈맥(torsades de pointes) 병력, 중증 저칼륨혈증, 중증 저마그네슘혈증이 있는 환자에게 이 약을 투여해서는 안 된다.
이 약은 QT 간격 연장이 발생할 유의한 위험이 있는 환자에게 주의해서 사용해야 한다. 이는 조절되지 않거나 유의한 심혈관 질환(예. 2도 또는 3도 심장차단(박동조율기 없이), 6개월 이내 심근경색, 조절되지 않는 협심증, 조절되지 않는 고혈압), 울혈성 심부전 병력이 있는 환자 및 QT 간격을 연장시키는 것으로 알려진 약물을 병용 투여한 환자를 포함한다. 전해질은 정상 범위로 유지되어야 한다 (‘용법용량’ 참조).
이 약의 치료 시작 전 ECGs 검사를 수행해야 하고, 전해질 이상이 있는 경우 투여 시작 전 교정하여야 한다. QTcF 간격이 450 ms 초과인 경우, 이 약 투여를 시작해서는 안 된다.
유도 및 공고요법 중, ECGs 검사는 투여 시작 전 수행하고, 이후에는 이 약 투여 중 주 1회 또는 임상적 증상에 따라 더 자주 수행해야 한다.
유지요법 중, ECGs 검사는 투여 시작 전 수행하고, 투여 시작 및 용량 증가 후 처음 1개월 동안에는 주 1회 수행해야 하며, 그 후에는 임상적 증상에 따라 수행해야 한다. QTcF 간격이 450 ms 초과인 경우 유지 시작 용량을 증가해서는 안 된다(표 1 참고).
생명을 위협하는 부정맥의 징후 또는 증상과 함께 QT 간격 연장이 나타난 환자에 대해서는 이 약을 영구 중단한다(‘용법 ▪용량’ 참고).
QT 간격의 ECG 모니터링은 QT 간격 연장 및 염전성 심실 빈맥(torsades de pointes)이 발생할 위험이 유의하게 있는 환자에서 더 자주 수행되어야 한다.
이 약의 치료 시작 전 및 치료 중에 저칼륨혈증 및 저마그네슘혈증의 모니터링 및 교정을 수행해야 한다. 설사나 구토를 경험한 환자에서는 전해질 및 심전도를 더 자주 모니터링 해야 한다.
⓵ QT 간격을 연장하는 약물 투여 시 ECG 모니터링
이 약과 QT 간격을 연장하는 것으로 알려진 약물을 함께 투여하는 경우, 환자의 ECG를 더 자주 모니터링 해야 한다. QT 간격을 연장하는 약물의 예는 아졸계 항진균제, 온단세트론, 그라니세트론, 아지스로마이신, 펜타미딘, 독시사이클린, 목시플록사신, 아토바쿠온, 프로클로르페라진 및 타크로리무스가 포함되지만 이에 국한되지 않는다.
⓶ 강력한 CYP3A 억제제와 병용 투여
이 약과 강력한 CYP3A 억제제를 병용 투여하는 경우, 퀴자티닙의 노출을 증가시킬 수도 있으므로 이 약의 용량을 감소해야 한다 (‘용법 ▪용량’ 참고).
2) 배 ▪태아 독성
동물 시험 결과에 근거하여, 이 약은 임부에게 투여 시 배 ▪태아에 위해할 수 있다. 임부에게 태아에 대한 잠재적 위험에 대해 알린다. 임신 가능성이 있는 여성에게 이 약 투여 중 및 마지막 투여 이후 최소 7개월 동안 효과적인 피임법을 사용해야 한다. 임신 가능성이 있는 배우자가 있는 남성은 이 약 투여 중 및 마지막 투여 이후 최소 4개월 동안 효과적인 피임법을 사용해야 한다.(‘7. 임부 및 수유부에 대한 투여’항 참고).
4. 이상반응
1) 안전성 프로파일 요약
이 약의 안전성은 FLT3 ‑ITD 양성인 새로 진단받은 급성 골수성 백혈병 성인 환자를 대상으로 한 무작위배정, 이중 눈가림, 위약 대조 임상시험(QuANTUM ‑First)에서 조사되었다. 환자들은 이 약(N=265) 또는 위약(N=268)을 표준 화학요법과 병용 투여받거나 단독 유지요법으로 투여받았다(‘14. 전문가를 위한 정보’항 참고). 전체 치료 기간의 중앙값은 이 약 치료군의 환자에 대해서는 10.7주(범위: 0.1 ~ 184.1)였고 위약 치료군의 환자에 대해서는 9.5주(범위: 0.4 ~ 181.9)였다.
전체 시험기간 중, 이 약 치료군에서 가장 흔하게 보고된 이상반응(발생률 >20%)은 알라닌 아미노 전이 효소 증가(58.9%), 혈소판 수 감소(40.0%), 헤모글로빈 감소(37.4%), 설사(37.0%), 구역(34.0%), 복통(29.4%), 두통(27.5%), 구토(24.5%) 및 중성구 수 감소(21.9%)였다. 가장 흔하게 보고된 3등급 또는 4등급 이상반응(발생률 ≧5%)은 혈소판 수 감소(40.0%), 헤모글로빈 감소(35.5%), 중성구 수 감소(21.5%), 알라닌 아미노 전이 효소 증가(12.1%) 균혈증(7.2%) 및 진균 감염(5.7%)이었다.
이 약 치료군에서 가장 흔하게 보고된 중대한 이상반응(발생률 ≧2%)은 중성구 감소증(3.0%), 진균 감염(2.3%) 및 헤르페스 감염(2.3%)이었다. 치명적인 결과를 나타낸 이상반응은 진균 감염(0.8%) 및 심정지(0.4%)였다.
이 약의 투여 일시 중단과 연관된 가장 흔하게 보고된 이상반응(발생률 ≧2%)은 중성구 감소증(10.6%), 혈소판 감소증(4.5%) 및 심전도 QT 연장(2.6%)이었다. 이 약의 용량 감소와 연관된 가장 흔하게 보고된 이상반응(발생률 ≧2%)은 중성구 감소증(9.1%), 혈소판 감소증(4.5%) 및 심전도 QT 연장(3.8%)이었다.
이 약의 영구 중단과 연관된 가장 흔하게 보고된 이상반응(발생률 ≧1%)은 혈소판 감소증(1.1%)이었다.
단독 유지요법 중 안전성 프로파일
유지 단계에 진입한 208명(39%)의 환자는 이 약(116명) 또는 위약(92명)을 투여받았다. 유지 요법을 위한 치료기간의 중앙값은 이 약 치료군의 환자에 대해서는 67.4주(범위: 0.4 ~ 167.0)였고 위약 치료군의 환자에 대해서는 67.7주(범위: 0.3 ~ 167.4)였다.
이 약 치료군에서 가장 흔하게 보고된 이상반응은 중성구 수 감소(87.9%), 혈소판 수 감소(72.4%), 헤모글로빈 감소(55.2%), 알라닌 아미노 전이 효소 증가(49.1%), 상기도 감염(27.6%), 구역(23.3%) 및 설사(20.7%)였다. 이 약 투여 시 가장 빈번하게 보고된 3등급 또는 4등급 이상반응은 중성구 수 감소(71.6%), 혈소판 수 감소(24.1%) 및 헤모글로빈 감소(18.1%)였다.
이 약 치료군에서 가장 흔하게 보고된 중대한 이상반응은 헤르페스 감염(3.4%), 중성구 감소증(2.6%), 상기도 감염(1.7%) 및 구토(1.7%)였다. 유지 요법 중 치명적인 이상반응은 발생하지 않았다.
이 약의 투여 일시 중단과 연관된 가장 흔하게 보고된 이상반응은 중성구 감소증(24.1%), 혈소판 감소증(10.3%) 및 심전도 QT 연장(4.3%)이었다. 이 약의 용량 감소와 연관된 가장 흔하게 보고된 이상반응은 중성구 감소증(20.7%), 혈소판 감소증(10.3%) 및 심전도 QT 연장(5.2%)이었다. 이 약의 영구 중단과 연관된 가장 흔하게 보고된 이상반응은 혈소판 감소증(2.6%), 구역, 중성구 감소증 및 식욕 감소(각각 1.7%)였다.
2) 이상반응 표
이상반응은 MedDRA 기관계 대분류(SOC)에 따라 기재되었다. 각 기관계 내에서, 이상반응은 다음의 통상의 규칙(CIOMS III)을 이용하여 가장 빈도가 높은 반응 순으로 빈도별로 정렬하였다: 매우 흔하게( ≧1/10), 흔하게( ≧1/100 ~ <1/10), 흔하지 않게( ≧1/1000 ~ <1/100), 드물게( ≧1/10,000 ~ <1/1000), 매우 드물게(<1/10,000). 각 빈도 범주 내에서 이상반응은 중증도가 감소하는 순서대로 기재하였다.
표 4: 이 약의 임상시험(QuANTUM ‑First)에서 관찰된 이상반응
|
약물이상반응 |
이 약 + 표준 화학요법 N=265 |
위약 + 표준 화학요법 N=268 | ||
|
모든 등급 (%) |
3등급 또는 4등급 (%) |
모든 등급 (%) |
3등급 또는 4등급 (%) | |
|
감염 및 침습 | ||||
|
매우 흔하게 | ||||
|
상기도 감염a |
18.1 |
1.9 |
10.1 |
2.2 |
|
진균 감염b |
15.1 |
5.7 |
9.7 |
3.0 |
|
헤르페스 감염c |
14.0 |
3.0 |
8.2 |
1.5 |
|
균혈증d |
11.3 |
7.2 |
4.1 |
3.7 |
|
혈액 및 림프계 이상 | ||||
|
흔하게 | ||||
|
범혈구 감소증 |
2.6 |
2.3 |
0.4 |
0.4 |
|
매우 흔하게 | ||||
|
혈소판 감소증e |
40.0 |
40.0 |
44.0 |
44.0 |
|
빈혈e |
37.4 |
35.5 |
45.5 |
42.5 |
|
중성구 감소증e |
21.9 |
21.5 |
21.6 |
20.1 |
|
대사 및 영양 이상 | ||||
|
매우 흔하게 | ||||
|
식욕 감소 |
17.4 |
4.9 |
13.4 |
1.9 |
|
신경계 이상 | ||||
|
매우 흔하게 | ||||
|
두통f |
27.5 |
0 |
19.8 |
0.7 |
|
심장 이상 | ||||
|
흔하지 않게 | ||||
|
심정지g |
0.8 |
0.4 |
0 |
0 |
|
심실세동g |
0.4 |
0.4 |
0 |
0 |
|
호흡기, 흉곽 및 종격 이상 | ||||
|
매우 흔하게 | ||||
|
비출혈 |
15.1 |
1.1 |
10.8 |
0.4 |
|
위장관 이상 | ||||
|
매우 흔하게 | ||||
|
설사h |
37.0 |
3.8 |
35.1 |
3.7 |
|
구역 |
34.0 |
1.5 |
31.3 |
1.9 |
|
복통i |
29.4 |
2.3 |
22.0 |
1.1 |
|
구토 |
24.5 |
0 |
19.8 |
1.5 |
|
소화 불량 |
11.3 |
0.4 |
8.6 |
0.7 |
|
간담도계 이상 | ||||
|
매우 흔하게 | ||||
|
알라닌 아미노 전이 효소 증가e |
58.9 |
12.1 |
60.1 |
10.4 |
|
전신 이상 및 투여 부위 상태 | ||||
|
매우 흔하게 | ||||
|
부종j |
18.9 |
0.4 |
19.0 |
1.5 |
|
임상 검사 | ||||
|
매우 흔하게 | ||||
|
심전도 QT 연장k |
14.0 |
3.0 |
4.1 |
1.1 |
|
표준 화학요법 = 시타라빈(사이토신 아라비노시드) 및 안트라사이클린(다우노루비신 또는 이다루비신) a 상기도 감염은 상기도 감염, 비인두염, 부비동염, 비염, 편도염, 인후두염, 세균성 인두염, 인두 편도염, 바이러스 인두염 및 급성 부비동염을 포함함. b 진균 감염은 구강 칸디다증, 기관지 폐 아스페르길루스증, 진균 감염, 외음질 칸디다증, 아스페르길루스 감염, 진균성 하기도 감염, 진균성 구강 감염, 칸디다 감염, 진균성 피부 감염, 털곰팡이증, 구강 인두 칸디다증, 구강 아스페르길루스증, 진균성 간 감염, 간 비장 칸디다증, 손발톱 진균증, 진균 혈증, 전신 칸디다 및 전신 진균증을 포함함. c 헤르페스 감염은 구강 헤르페스, 대상 포진, 헤르페스 바이러스 감염, 단순 포진, 사람 헤르페스 바이러스 6형 감염, 생식기 헤르페스 및 헤르페스 피부염을 포함함. d 균혈증은 균혈증, 클레브시엘라 균혈증, 포도상 구균 균혈증, 장구균성 균혈증, 연쇄구균 균혈증, 기기 관련 균혈증, 대장균 균혈증, 코리네박테륨 균혈증 및 슈도모나스 균혈증을 포함함. e 용어들은 실험실 데이터를 기반으로 한다. f 두통은 두통, 긴장성 두통 및 편두통을 포함함. g 1명이 2건의 사례(심실세동 및 심정지)를 경험하였다. h 설사는 설사 및 출혈성 설사를 포함함. i 복통은 복통, 상복부 통증, 복부 불편감, 하복부 통증 및 위장관 통증을 포함함. j 부종은 말초 부종, 안면 부종, 부종, 체액 과부하, 전신 부종, 말초 종창, 국소 부종 및 얼굴 종창을 포함함. k 심전도 QT 연장은 심전도 QT 연장 및 심전도 QT 간격 이상을 포함함. | ||||
이 약은 ECG에서 QT 간격을 연장한다. 등급에 관계없이, 치료 후 발생한 이상반응으로써 QT 간격 연장은 이 약을 투여한 환자의 14.0%에서 보고되었고, 환자의 3.0%가 3등급 이상의 이상반응을 경험하였다. QT 연장은 환자 10명(3.8%)에서는 용량 감소, 7명(2.6%)에서는 투여 일시 중단, 2명(0.8%)에서는 영구 중단을 초래하였다. 중앙에서 검토된 ECG 데이터에 근거하여, QTcF >500 ms는 환자의 2.3%에서 발생하였다. 이 약을 투여한 환자 2명(0.8%)이 심실세동 기록과 함께 심정지를 경험하였고, 1명은 치명적인 결과를 나타냈으며, 이들 모두 중증 저칼륨혈증이 있는 상황에서 발생하였다. 이 약 투여 전 및 투여 중 심전도 검사 및 모니터링, 저칼륨혈증 및 저마그네슘혈증 교정을 실시해야 한다. QT 간격 연장이 있는 환자에 대한 이 약의 용량 조절은 ‘용법 ▪용량’ 항을 참고한다.
5. 일반적 주의
1) FLT3 ‑ITD 변이 검사
이 약은 FLT3 ‑ITD 변이 양성인 급성 골수성 백혈병 환자에서만 치료 이익을 보였으므로, 이 약의 치료를 받을 환자를 선택하기 위해서는 FLT3 ‑ITD 변이 양성인 급성 골수성 백혈병의 진단이 필요하다. FLT3 ‑ITD 상태는 임상시험 분석법(PCR과 모세관 전기영동법)을 사용하여 전향적으로 확인되었고, FDA에서 승인받은 분석법인 LeukoStratⓡ CDx FLT3 분석 동반진단기기를 사용하여 후향적으로 검증되었다.
6. 약물 상호작용
1) 강력한 CYP3A 억제제
건강한 대상자에게 이 약과 강력한 CYP3A 억제제와 병용투여 시, 이 약 단독 투여와 비교하여 퀴자티닙의 노출이 증가하였다. 이 약의 1회 투여 용량(26.5 mg)과 케토코나졸(200 mg씩 1일 2회)을 병용투여 시 퀴자티닙의 Cmax는 17 %, AUCinf는 94 % 증가하였다. 활성 대사체인 AC886의 AUCinf는 15 %, Cmax는 60 % 감소했다. 정상 상태에서 퀴자티닙의 노출(Cmax 및 AUC0 ‑24h)은 각각 86 %, 96 % 증가할 것으로 추정되었다. 퀴자티닙의 노출 증가로 독성의 위험성이 증가할 수도 있다.
강력한 CYP3A 억제제와의 병용투여가 불가피한 경우, 이 약의 용량을 감소해야 한다(‘용법용량’ 참고). 강력한 CYP3A 억제제의 예로 케토코나졸, 이트라코나졸, 포사코나졸, 보리코나졸, 클래리스로마이신, 네파조돈, 텔리스로마이신 및 항레트로바이러스제가 있다.
2) 강력한 및 중등도의 CYP3A 유도제
건강한 대상자에게 이 약과 중등도의 CYP3A 유도제의 병용투여 시, 이 약 단독 투여(총 53 mg)와 비교하여 퀴자티닙 및 활성 대사체 AC886의 노출이 감소하였다. 중등도의 CYP3A 유도제인 에파비렌즈(600 mg 씩 1일 1회)와 병용투여 시 퀴자티닙 Cmax 및 AUCinf는 각각 약 45 %와 90 % 감소하였다. AC886의 Cmax 및 AUCinf는 각각 약 68 % 및 96 % 감소하였다 (‘14. 전문가용 정보’항 참고).
퀴자티닙 노출 감소는 유효성 감소로 이어질 수 있다. 이 약과 강력한 또는 중등도의 CYP3A 유도제와의 병용투여를 피해야 한다.
강력한 CYP3A4 유도제의 예로는 아팔루타마이드, 카르바마제핀, 엔잘루타마이드, 미토탄, 페니토인, 리팜피신, 세인트 존스 워트(Hypericum perforatum 라고도 함) 등 특정 생약 제제가 있다. 중등도의 CYP3A4 유도제의 예로는 에파비렌즈, 보젠탄, 에트라비린, 페노바르비탈 및 프리미돈이 있다.
3) 수송체
⓵ P ‑당단백(P ‑gp) 기질
퀴자티닙과 다비가트란 에텍실레이트(P ‑gp 기질)를 병용투여하였을 때, 총 및 유리 다비가트란의 Cmax는 각각 12% 및 13% 증가하였고, AUCinf는 각각 13% 및 11% 증가하였다(‘14. 전문가용 정보’항 참고). 퀴자티닙은 약한 P ‑gp 억제제이며, 이 약과 P ‑gp 기질을 병용투여 시 용량 조절은 권장되지 않는다.
⓶ 유방암 내성 단백질(BCRP) 기질
퀴자티닙은 in vitro IC50 추정값 0.813 μM에서 유방암 내성 단백질을 저해한다. 생리학적 기반 약동학(PBPK) 분석에 따르면 퀴자티닙 53mg은 로수바스타틴의 AUC0 ‑48h를 2.44배, 설파살라진은 2.66배 증가시킬 것으로 예측되었다. 퀴자티닙을 BCRP 기질 약물과 병용투여할 경우 주의해야 한다. BCRP 기질 약물의 용량 감소를 고려해야 한다.
⓷ Uridine Diphosphate Glucuronosyltransferases(UGT) 1A1 기질
퀴자티닙은 in vitro Ki 추정값 0.78 μM에서 UGT1A1을 저해한다. PBPK 분석에 근거하여, 퀴자티닙은 랄테그라비르(UGT1A1 기질)의 Cmax와 AUCinf를 3% 증가시킬 것으로 예상되었다. UGT1A1 기질과 이 약의 병용투여 시 용량 조절은 권장되지 않는다.
⓸ QT 간격을 연장시키는 약물
이 약과 QT 간격을 연장하는 기타 약물의 병용투여는 QT 연장의 발생률을 더욱 증가시킬 수 있다. 이 약과 QT 간격을 연장하는 약물을 병용투여 시 주의해야 한다(‘3. 다음 환자에는 신중히 투여할 것 중 ‘1) QT 간격 연장’항 참고).
7. 임부 및 수유부에 대한 투여
1) 남성 및 여성의 피임
이 약은 임부에게 투여 시 배 ▪태아에 해를 끼칠 수 있다(‘14. 전문가용 정보’항 참고). 잠재적인 임신 가능성이 있는 여성은 이 약 투여 중 및 마지막 투여 후 최소 7개월 동안 효과적인 피임법을 사용하여야 한다. 임신 가능성이 있는 여성 배우자가 있는 남성은 이 약 투여 중 및 마지막 투여 후 최소 4개월 동안 효과적인 피임법을 사용하여야 한다.
2) 임부
임부에게 퀴자티닙을 투여한 자료는 없다. 동물시험 결과에 근거하여, 이 약은 임부에게 투여 시 배 ▪태아 독성을 초래할 수 있다 (‘14. 전문가용 정보’항 참고).
이 약을 임신 중 투여해서는 안 된다. 임부에게 태아에 대한 잠재적 위험에 대해 알린다.
3) 수유부
퀴자티닙 또는 활성 대사체가 모유로 분비되는지 여부는 알려져 있지 않다. 모유 수유 중인 소아에서 중대한 이상반응이 발생할 가능성이 있기 때문에, 이 약 투여 중 및 마지막 투여 후 최소 5주 동안 모유 수유를 해서는 안 된다.
4) 임신 가능성이 있는 여성
임신 가능성이 있는 여성은 이 약으로 치료를 시작하기 전 임신 검사를 받아야 한다.
5) 생식능
이 약이 사람의 생식능에 미치는 영향에 대한 자료는 없다. 동물 시험 결과에 근거할 때, 이 약 투여로 인해 여성 및 남성 생식능이 손상될 수 있다(‘14. 전문가를 위한 정보’항 참고).
8. 소아에 대한 투여
이 약은 18세 미만의 소아 및 청소년에서 안전성 및 유효성이 확립되지 않았다. 가용할 수 있는 데이터가 없다.
9. 고령자에 대한 투여
65세 이상 환자에서 이 약의 용량 조절은 필요하지 않다.
임상시험(QuANTUM ‑First)에서 이 약을 투여받은 새로 진단받은 급성 골수성 백혈병 환자 265명 중 69명(26%)이 65세 이상이었고 1명(0.4%)이 75세였다. 65세 이상 환자와 젊은 성인 환자 간에 안전성에서 전반적인 차이는 관찰되지 않았다.
이 약의 임상시험에는 젊은 성인 환자와 다르게 반응을 보이는지를 확인하기에 충분한 수의 65세 이상 환자가 포함되지 않았다.
이 약은 특히 치료 초기 단계에서 젊은 환자에 비해 고령 환자(즉, 65세 이상)에서 치명적인 감염 발생 빈도가 더 높았다. 65세 이상 환자는 유도 요법 기간 동안 중증 감염 발생 여부를 면밀히 모니터링해야 한다.
10. 신 장애 환자에 대한 투여
경증 또는 중등도 신 장애 환자에서 용량 조절은 권장되지 않는다.
이 약은 중증 신 장애(Cockcroft ‑Gault 식으로 추정한 CLcr <30 mL/min) 환자에서 안전성 및 유효성이 평가되지 않았기 때문에, 중증 신 장애 환자에게는 권장되지 않는다.
11. 간 장애 환자에 대한 투여
경증 또는 중등도 간 장애 환자에서 용량 조절은 권장되지 않는다.
이 약은 중증 간 장애(Child ‑Pugh Class C 또는 총 빌리루빈 >3 x ULN 및 모든 수치의 AST) 환자에서 안전성 및 유효성이 평가되지 않았기 때문에, 중증 간 장애 환자에게는 권장되지 않는다.
12. 과량투여시의 처치
이 약의 과량투여에 대한 해독제는 알려지지 않았다. 과량투여 시 투여를 일시 중단하고, QT 간격 연장을 측정하기 위한 ECG를 수행하며, 적절한 보조 조치를 한다(‘용법용량’ 및 ‘3. 신중투여’항 참고).
13. 보관 및 취급 시 주의사항
1) 이 약은 어린의 손이 닿지 않는 곳에 보관해야 한다.
2) 이 약을 다른 용기에 옮겨 담지 않는다.
14. 전문가를 위한 정보
1) 약리작용
(1) 작용 기전
퀴자티닙은 티로신 키나아제 FLT3 수용체의 저분자 억제제다. 퀴자티닙 및 그 주요 순환 활성 대사체 AC886은 FLT3의 아데노신삼인산(ATP) 결합 부위에 높은 친화성(각각 Kd = 1.3 nM 및 0.54 nM)으로 경쟁적으로 결합한다. 퀴자티닙 및 AC886은 수용체의 자가인산화를 방지하여 FLT3 키나아제 활성을 억제하며, 이후 다운스트림 FLT3 수용체 신호전달을 억제하고 FLT3 ‑ITD 의존적인 세포 증식을 차단한다.
(2) 약력학적 효과
심장 전기생리학
임상시험(QuANTUM ‑First)의 노출 ‑반응 분석에서 유지요법 중 퀴자티닙(53 mg)의 정상 상태 Cmax에서 연장된 QTcF 간격은 24.1 ms(양측 90% CI의 상한: 26.6 ms)였으며 농도 ‑의존적인 QTcF 간격 연장이 예상되었다.
2) 약동학적 특성
퀴자티닙 및 주요 순환 활성 대사체(AC886)의 약동학은 건강한 성인과 성인 암 환자에서 분석되었으며, 달리 명시되지 않는 한 기하 평균(변동계수 [%CV])으로 제시되었다.
급성 골수성 백혈병 성인 환자에서 1일 1회 투여 후 15일 째에 퀴자티닙 농도가 정상 상태(steady state)에 도달했다. 건강한 성인의 경우 퀴자티닙 노출(AUC 및 Cmax)은 26.5 mg (권장 유도 용량 35.4 mg의 0.75배)에서 79.5 mg (권장 유도 용량 35.4 mg의 2.25배)까지, 급성 골수성 백혈병 성인 환자의 경우 17.7mg (권장 유도 용량 35.4mg의 0.5배)에서 53 mg (권장 유도 용량 35.4 mg의 1.5배)까지의 용량 범위에서 비례적으로 증가했다.
표 5. 퀴자티닙 및 AC886의 약동학적 특성
|
|
퀴자티닙 |
AC886 |
|
새로 진단된 급성 골수성 백혈병 환자의 유도 요법 중 이 약 35.4mg을 1일 1회 투여한 후 정상 상태 노출도 | ||
|
Cmax |
140 ng/mL (71%) |
163 ng/mL (52%) |
|
AUC0-24h |
2,680 ng·h/mL (85%) |
3,590 ng·h/mL (51%) |
|
새로 진단된 급성 골수성 백혈병 환자의 공고 요법 중 이 약 35.4mg을 1일 1회 투여한 후 정상 상태 노출도 | ||
|
Cmax |
204 ng/mL (64%) |
172 ng/mL (47%) |
|
AUC0-24h |
3,930 ng·h/mL (78%) |
3,800 ng·h/mL (46%) |
|
새로 진단된 급성 골수성 백혈병 환자의 유지 요법 중 이 약 53mg을 1일 1회 투여한 후 정상 상태 노출도 | ||
|
Cmax |
529 ng/mL (60%) |
262 ng/mL (48%) |
|
AUC0-24h |
10,200 ng·h/mL (75%) |
5,790 ng·h/mL (46%) |
|
축적비* (AUC0-24h) |
5.4 (4.4) |
8.7 (6.8) |
(1) 흡수
정제 제형에서 퀴자티닙의 절대 생체이용률은 71 %였다. 건강한 성인에게 공복 상태에서 경구 투여 시 퀴자티닙 및 AC886의 최고 농도 도달 시간(Tmax 중앙값)은 각각 약 4시간(범위 2 ‑ 8시간) 및 5 ‑ 6시간(범위 4 ‑ 120시간)이었다. 53 mg/일 용량으로 유지요법 중 정상 상태에서, 퀴자티닙 및 AC886의 기하평균(%CV) Cmax는 각각 529 ng/mL(60 %) 및 262 ng/mL(48 %)로 추정되었고, 기하평균(%CV) AUC0 ‑24h는 각각 10,200 ng ▪h/mL(75 %) 및 5,790 ng ▪h/mL(46 %)였다.
음식과 함께 이 약 투여 시 퀴자티닙 Cmax는 8% 감소하였으며, AUCinf는 8% 증가하였다. Tmax는 2시간 지연되었다. 이 약은 음식 여부와 관계없이 투여 가능하다.
(2) 분포
퀴자티닙 및 AC886의 In vitro 사람 혈장 단백질 결합률은 각각 99% 이상이다.
10 ‑ 1,000 ng/mL 농도 범위에서 in vitro 적혈구 내 퀴자티닙 및 AC886의 혈액 대 혈장 비율은 각각 0.79 ‑ 1.30 및 1.36 ‑ 3.19이다.
건강한 성인에서 퀴자티닙의 정상 상태에서 분포 용적(Vss)의 기하평균(%CV)은 275 L(17%)로 추정되었다.
(3) 생체 내 변환
퀴자티닙은 활성 대사체 AC886을 생성하는 산화 경로를 통해 in vitro에서 CYP3A4/5에 의해 주로 대사되며, 이후 CYP3A4/5에 의해 추가로 대사된다. 유지요법 중 정상 상태에서 퀴자티닙에 대한 AC886의 AUC0 ‑24h비는 0.57이었다.
(4) 배설
퀴자티닙 및 AC886의 유효 반감기(t1/2)의 평균(표준편차)는 새로 진단된 급성 골수성 백혈병 환자에서 각각 81시간(±73) 및 136시간(±113)이다. 퀴자티닙 및 AC886의 축적비의 평균 AUC0 ‑24h(표준편차)는 각각 5.4(±4.4) 및 8.7(±6.8)이었다.
퀴자티닙 및 그 대사체는 대부분이 일차적으로 간담도 경로를 통해 제거되어 대변으로 배설된다(경구 투여된 방사성 동위원소 표지 투여량의 76.3 %). 대변으로 배설되는 미변화체 퀴자티닙은 경구 투여된 방사성 동위원소 표지 투여량의 약 4 %였다. 신배설은 투여된 방사성 동위원소 표지량 제거의 부수적 경로이다(2 % 미만).
건강한 성인에서 퀴자티닙의 기하평균(%CV) 전신 청소율(CL)은 2.23 L/hour(29%)로 추정되었다.
(5) 선형성/비 ‑선형성
퀴자티닙은 건강한 성인에서 26.5 mg ~ 79.5 mg 용량 범위에서, 급성 골수성 백혈병 환자에서 17.7 mg ~ 53 mg 용량 범위에서 용량 비례적 선형성을 보였다.
3) 임상시험 정보
이 약의 유효성과 안전성은 FLT3 ‑ITD 변이 양성인 새로 진단받은 급성 골수성 백혈병 18 ‑ 75세 성인 환자(539명)를 대상으로 한 무작위배정, 이중 눈가림, 위약 대조, 3상 임상시험(QuANTUM ‑First)에서 평가되었다. FLT3 ‑ITD 상태는 임상시험 분석법(PCR과 모세관 전기영동법)을 사용하여 전향적으로 확인되었고, FDA에서 승인받은 분석법인 LeukoStratⓡ CDx FLT3 분석 동반진단기기를 사용하여 후향적으로 검증되었다. 환자들은 표준 화학요법과의 병용요법(반응을 보인 환자의 경우 유도요법 후 공고요법) 주기마다 2주간(유도요법 시 8 ‑21일, 공고요법 시 6 ‑19일) 이 약 35.4 mg(n=268) 또는 위약(n=271)을 1일 1회 투여한 후, 이 약(2주간 1일 1회 26.5 mg을 투여하고 이후에는 1일 1회 53 mg을 투여) 또는 위약 단독 유지요법을 최대 36주기(28일/1주기) 동안 투여받도록 무작위배정되었다 (1:1).
환자들은 최대 2주기의 유도 화학요법(다우노루비신 [제1일, 제2일, 제3일에 60 mg/m2/일 IV] 또는 이다루비신 [제1일, 제2일, 제3일에 12 mg/m2/일 IV]과 시타라빈 [7일 동안 100 mg/m2/일 또는 시험기관 또는 현지 표준에 따라 허용되는 경우 200 mg/m2/일, IV 주입]) 후, 최대 4주기의 공고 화학요법 및/또는 HSCT로 구성된 관해 후 치료를 받았다. 공고 화학요법은 60세 미만 환자에 대해 3.0 g/m2 및 60세 이상 환자에 대해 1.5 g/m2의 시타라빈을 제1일, 제3일, 제5일에 12시간마다 IV 주입으로 투여하였다. HSCT를 진행한 환자들은 전처치 시작 7일 전에 시험 치료를 중단하였다.
무작위배정된 두 치료군은 베이스라인 인구통계학적 특성, 질병 특성 및 층화 요인과 관련하여 적절하게 균형을 이루었다. 무작위배정된 539명의 연령 중앙값은 56세(범위 20 ‑75세)였고, 54.5%는 여성, 45.5%는 남성, 59.7%는 백인, 29.3%는 아시아인, 1.3%는 흑인 또는 아프리카계 미국인, 9.7%는 기타 인종이었다. 84%에서 베이스라인 ECOG 전신수행상태(PS) 점수가 0 또는 1이었다. 대다수의 환자(72.4%)가 베이스라인에서 세포유전학적 위험이 중간 상태였다. FLT3 ‑ITD 변이형 대립유전자 빈도(VAF)는 환자의 35.6%에서 3 ‑25%였고, 환자의 52.1%에서는 >25 ‑50%이었고, 환자의 12.1%에서 >50%이었다. 환자의 52%에서 NPM1 변이가 확인되었다.
1차 유효성 평가변수는 무작위 배정일부터 모든 원인에 의한 사망까지의 전체 생존(OS)이었다.
1차 분석은 마지막 환자의 무작위 배정 후 최소 24개월의 추적 관찰 후 수행하였으며 이 약 투여군에서 위약 투여군 대비 전체 생존기간이 통계적으로 유의하게 개선되었다 [위험비(HR) 0.78; 95% CI: 0.62, 0.98; 양측 p=0.0324] (그림 1 참조). 추적관찰 기간의 중앙값은 39.2개월이었다.
이 약과 표준 화학요법과의 병용투여군에서는 유도요법과 공고요법을 각각 65 %, 44 %의 환자가 완료하였고, 위약과 표준 화학요법과의 병용투여군에서는 각각 65 %, 34 %의 환자가 완료하였다. 유지요법을 받은 환자는 208명(39%)이었고, 이 중 64%가 최소 12주기를 완료하였으며, 36%가 최소 24주기, 16%가 36주기를 완료하였다. 유지요법을 받은 208명 중 조혈모세포 이식 없이 공고요법 후 이 약 또는 위약으로 유지 요법을 받은 89명(43%)의 환자를 대상으로 한 탐색적 하위군 분석에서 전체 생존(OS) 위험비(HR)는 0.40 (95% CI: 0.19, 0.84)이었다. 208명 중 조혈모세포 이식 후 이 약 또는 위약으로 유지 요법을 받은 119명(57%)의 환자에서 OS 위험비는 1.62 (95% CI: 0.62, 4.22)이었다.
2차 평가변수인 무사건 생존은 통계적 유의성을 입증하지 못했다.
그림 1: QuANTUM ‑First에서 전체 생존에 대한 카플란 ‑마이어 곡선
4) 독성시험 정보
퀴자티닙을 사용한 발암성 시험은 수행되지 않았다.
유전독성 시험에서, 퀴자티닙은 박테리아 복귀돌연변이 시험에서 변이원성이었으나, 포유류 세포 돌연변이 분석(마우스 림프종 티미딘 키나아제) 또는 생체 내 형질전환 설치류 돌연변이 분석에서는 변이원성이 아니었다. 퀴자티닙은 사람 림프구 염색체 이상 시험 또는 생체 내 랫드 골수 소핵 시험에서 유전독성을 나타내지 않았다.
배 ▪태자 생식 독성 시험에서, 임신한 랫드에게 기관 형성 기간 동안 퀴자티닙을 경구 투여하였다. 6 mg/kg/일 (AUC 기반 53 mg/일인 권장 인체투여량[RHD]의 약 3배)를 투여하였을 때 태자 독성(낮은 태자 체중, 골격 골화에 대한 영향) 및 기형 유발성(부종을 포함한 태자 이상)이 관찰되었다. 10 mg/kg/일 초과의 용량에서, 배 ‑태자 치사성 및 착상 후 손실 증가가 관찰되었다. 모체 독성은 10 mg/kg/일 초과의 용량에서는 체중 증가 감소 및 사료 섭취량 감소로 나타났고, 20 mg/kg/일 용량에서는 이상 임상 징후로 나타났다.
퀴자티닙을 사용한 동물에서의 생식능 시험은 수행되지 않았다. 그러나, 랫드와 원숭이의 반복투여 독성시험에서 수컷 및 암컷 생식기관에 이상적 소견이 관찰되었다. 암컷 랫드의 경우, 10 mg/kg/일(AUC 기반 53 mg/일인 권장 인체투여량 [RHD]의 약 10배)에서 난소 낭종 및 질 점막 변형이 관찰되었다. 암컷 원숭이에서의 소견은 6 mg/kg/일(AUC 기반 RHD의 약 0.3배) 이상의 용량에서 자궁, 난소 및 질 위축이 관찰되었다. 수컷 랫드의 경우, 10 mg/kg/일(AUC 기반 RHD의 약 8배)의 용량에서 정소의 정세관 변성 및 정자 배출 장애가 관찰되었다. 수컷 원숭이에서의 소견은 6 mg/kg/일(AUC 기반 RHD의 약 0.5배) 이상의 용량에서 정소의 생식세포 감소가 관찰되었다. 4주 간의 회복기간 후, 암컷 랫드에서의 질 점막 변형을 제외한 모든 소견은 회복되었다.